Comparative Microbiome and Metabolome Analyses of the Marine Tunicate Ciona intestinalis from Native and Invaded Habitats

 , , , ,
, , , ,
Abstract
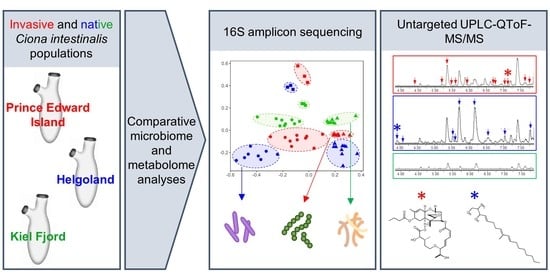
:
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Genotyping
2.3. Microbiome Analysis
2.3.1. DNA Extraction, Library Preparation, and Sequencing
2.3.2. Bioinformatic Processing and Statistical Analysis
2.4. Metabolome Analysis
2.4.1. Solvent Extraction
2.4.2. LC-MS/MS Analysis and Data Pre-Processing
2.4.3. Molecular Networking and Dereplication
2.4.4. Statistical Analysis
3. Results
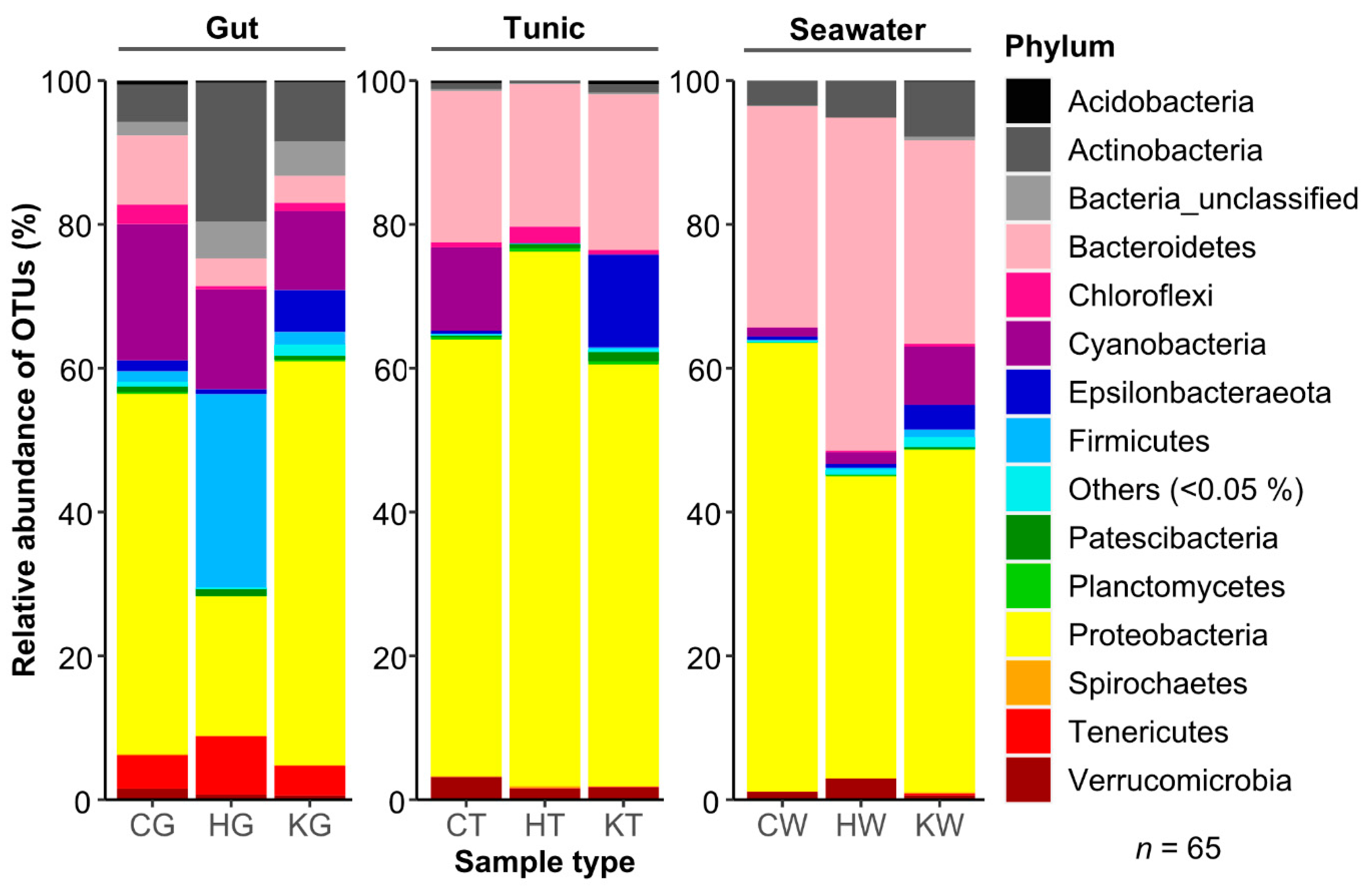
3.1. Comparative Microbiome Analysis of Tunic and Gut From Invasive and Native C. intestinalis
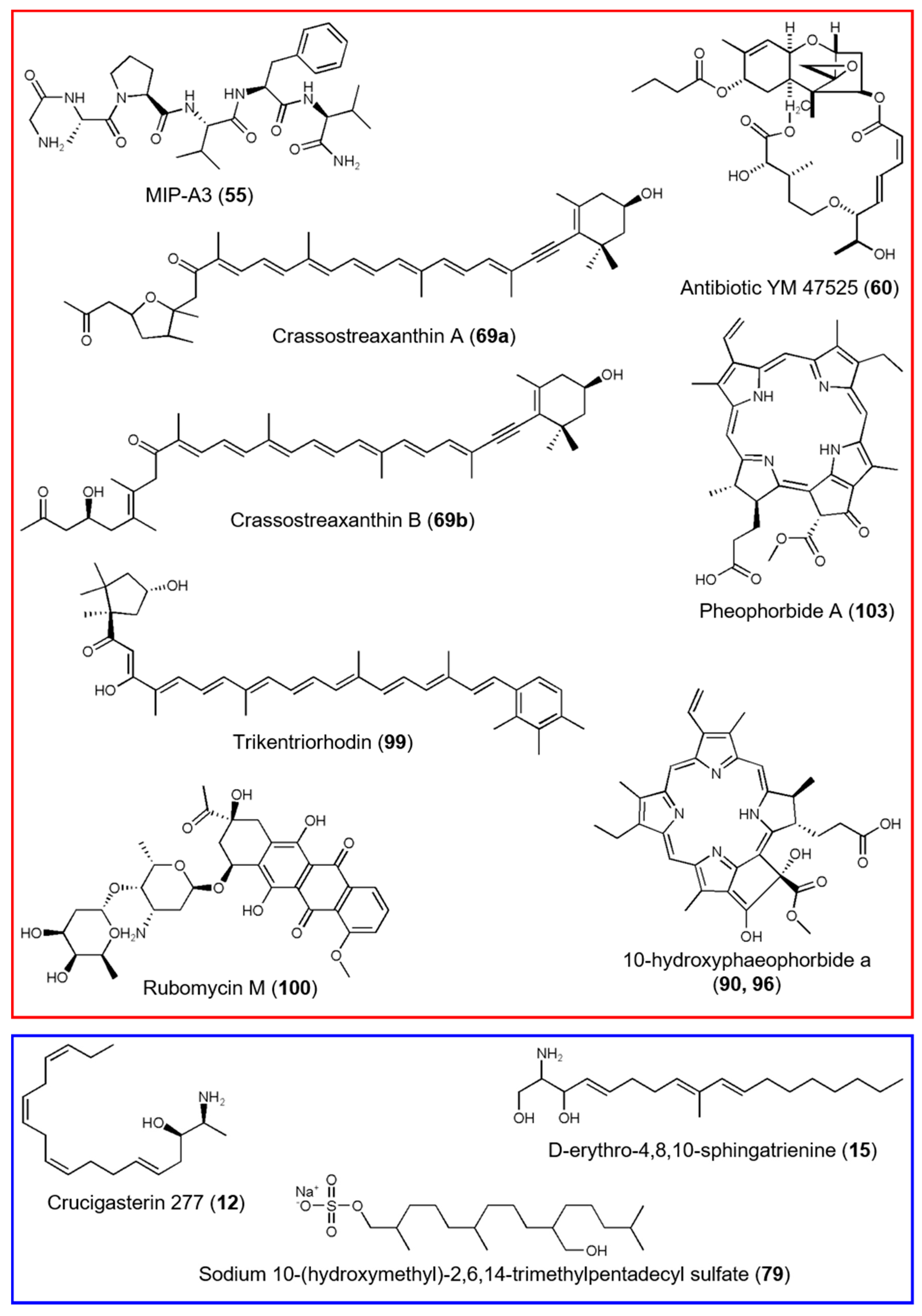
3.2. Comparative Metabolomics of C. intestinalis Extracts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keane, R.M.; Crawley, M.J. Exotic plant invasions and the enemy release hypothesis. Trends Ecol. Evol. 2002, 17, 164–170. [Google Scholar] [CrossRef]
- Blackburn, T.M.; Bellard, C.; Ricciardi, A. Alien versus native species as drivers of recent extinctions. Front. Ecol. Environ. 2019, 17, 203–207. [Google Scholar] [CrossRef]
- Simberloff, D.; Martin, J.L.; Genovesi, P.; Maris, V.; Wardle, D.A.; Aronson, J.; Courchamp, F.; Galil, B.; Garcia-Berthou, E.; Pascal, M.; et al. Impacts of biological invasions: What’s what and the way forward. Trends Ecol. Evol. 2013, 28, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, A.; Briski, E.; Bock, D.G.; Ghabooli, S.; MacIsaac, H.J. Ascidians as models for studying invasion success. Mar. Biol. 2015, 162, 2449–2470. [Google Scholar] [CrossRef]
- Molnar, J.L.; Gamboa, R.L.; Revenga, C.; Spalding, M.D. Assessing the global threat of invasive species to marine biodiversity. Front. Ecol. Environ. 2008, 6, 485–492. [Google Scholar] [CrossRef]
- Astudillo, J.C.; Leung, K.M.; Bonebrake, T.C. Seasonal heterogeneity provides a niche opportunity for ascidian invasion in subtropical marine communities. Mar. Environ. Res. 2016, 122, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.T.; Briski, E. An overview of recent research in marine biological invasions. Mar. Biol. 2017, 164, 121. [Google Scholar] [CrossRef] [Green Version]
- Doorduin, L.J.; Vrieling, K. A review of the phytochemical support for the shifting defence hypothesis. Phytochem. Rev. 2011, 10, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, N.; Rohde, S.; Hiromori, S.; Schupp, P.J. Understanding the invasion success of Sargassum muticum: Herbivore preferences for native and invasive Sargassum spp. Mar. Biol. 2016, 163, 181. [Google Scholar] [CrossRef]
- Callaway, R.M.; Ridenour, W.M. Novel weapons: Invasive success and the evolution of increased competitive ability. Front. Ecol. Environ. 2004, 2, 436–443. [Google Scholar] [CrossRef]
- Svensson, J.R.; Nylund, G.M.; Cervin, G.; Toth, G.B.; Pavia, H.; Callaway, R. Novel chemical weapon of an exotic macroalga inhibits recruitment of native competitors in the invaded range. J. Ecol. 2013, 101, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, K.P.; Bacon, C.; Bickford, W.; Braun, H.; Clay, K.; Leduc-Lapierre, M.; Lillard, E.; McCormick, M.K.; Nelson, E.; Torres, M.; et al. Advancing the science of microbial symbiosis to support invasive species management: A case study on Phragmites in the Great Lakes. Front. Microbiol. 2015, 6, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilcinskas, A. Pathogens as biological weapons of invasive species. PLoS Pathog. 2015, 11, e1004714. [Google Scholar] [CrossRef] [PubMed]
- Capinha, C.; Larson, E.R.; Tricarico, E.; Olden, J.D.; Gherardi, F. Effects of climate change, invasive species, and disease on the distribution of native European crayfishes. Conserv. Biol. 2013, 27, 731–740. [Google Scholar] [CrossRef]
- Holland, L.Z. Tunicates. Curr. Biol. 2016, 26, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Carver, C.E.; Mallet, A.L.; Vercaemer, B. Biological synopsis of the solitary tunicate Ciona intestinalis. Can. Man. Rep. Fish. Aquat. Sci. 2006, 2746, 1–55. [Google Scholar]
- Rius, M.; Heasman, K.G.; McQuaid, C.D. Long-term coexistence of non-indigenous species in aquaculture facilities. Mar. Pollut. Bull. 2011, 62, 2395–2403. [Google Scholar] [CrossRef]
- Bouchemousse, S.; Bishop, J.D.; Viard, F. Contrasting global genetic patterns in two biologically similar, widespread and invasive Ciona species (Tunicata, Ascidiacea). Sci. Rep. 2016, 6, 24875. [Google Scholar] [CrossRef] [Green Version]
- Blum, J.C.; Chang, A.L.; Liljesthrom, M.; Schenk, M.E.; Steinberg, M.K.; Ruiz, G.M. The non-native solitary ascidian Ciona intestinalis (L.) depresses species richness. J. Exp. Mar. Biol. Ecol. 2007, 342, 5–14. [Google Scholar] [CrossRef]
- Ramsay, A.; Davidson, J.; Landry, T.; Arsenault, G. Process of invasiveness among exotic tunicates in Prince Edward Island, Canada. Biol. Invasions 2008, 10, 1311–1316. [Google Scholar] [CrossRef]
- Daigle, R.M.; Herbinger, C.M. Ecological interactions between the vase tunicate (Ciona intestinalis) and the farmed blue mussel (Mytilus edulis) in Nova Scotia, Canada. Aust. J. Chem. 2009, 4, 177–187. [Google Scholar] [CrossRef]
- Patanasatienkul, T.; Sanchez, J.; Davidson, J.; Revie, C.W. The application of a mathematical model to evaluate the effectiveness of control strategies against Ciona intestinalis in mussel production. Front. Vet. Sci. 2019, 6, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, J.; Johannesson, K.; McQuaid, C.D.; Rius, M. Secondary contacts and genetic admixture shape colonization by an amphiatlantic epibenthic invertebrate. Evol. Appl. 2019, 13, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Ni, P.; Murphy, K.J.; Wyeth, R.C.; Bishop, C.D.; Li, S.; Zhan, A. Significant population methylation divergence and local environmental influence in an invasive ascidian Ciona intestinalis at fine geographical scales. Mar. Biol. 2019, 166, 143. [Google Scholar] [CrossRef]
- Evans, J.S.; Erwin, P.M.; Shenkar, N.; Lopez-Legentil, S. A comparison of prokaryotic symbiont communities in nonnative and native ascidians from reef and harbor habitats. FEMS Microbiol. Ecol. 2018, 94, fiy139. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Allard, P.M.; Peresse, T.; Bisson, J.; Gindro, K.; Marcourt, L.; Pham, V.C.; Roussi, F.; Litaudon, M.; Wolfender, J.L. Integration of molecular networking and in-silico MS/MS fragmentation for natural products dereplication. Anal. Chem. 2016, 88, 3317–3323. [Google Scholar] [CrossRef]
- Parrot, D.; Blümel, M.; Utermann, C.; Chianese, G.; Krause, S.; Kovalev, A.; Gorb, S.N.; Tasdemir, D. Mapping the surface microbiome and metabolome of brown seaweed Fucus vesiculosus by amplicon sequencing, integrated metabolomics and imaging techniques. Sci. Rep. 2019, 9, 1061. [Google Scholar] [CrossRef]
- Iannelli, F.; Pesole, G.; Sordino, P.; Gissi, C. Mitogenomics reveals two cryptic species in Ciona intestinalis. Trends Genet. 2007, 23, 419–422. [Google Scholar] [CrossRef] [Green Version]
- Waters, J.M.; Dijkstra, L.H.; Wallis, G.P. Biogeography of a southern hemisphere freshwater fish: How important is marine dispersal? Mol. Ecol. 2000, 9, 1815–1821. [Google Scholar] [CrossRef]
- Zhan, A.; Macisaac, H.J.; Cristescu, M.E. Invasion genetics of the Ciona intestinalis species complex: From regional endemism to global homogeneity. Mol. Ecol. 2010, 19, 4678–4694. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal-W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Lee, C.; Kim, J.; Hwang, S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 2005, 89, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bushnell, B. BBTools Software Package. 2019. Available online: http://sourceforge.net/projects/bbmap (accessed on 30 June 2019).
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Huse, S.M.; Welch, D.M.; Morrison, H.G.; Sogin, M.L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 2010, 12, 1889–1898. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Enviroment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Package vegan v. 2.5-6: Community ecology package. 2019. Available online: https://cran.r-project.org/src/contrib/Archive/vegan/ (accessed on 14 January 2020).
- Kembel, S.W.; Cowan, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, 180–188. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasiak, L.C.; Zinder, S.H.; Buckley, D.H.; Hill, R.T. Bacterial diversity associated with the tunic of the model chordate Ciona intestinalis. ISME J. 2014, 8, 309–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, P.L.; Fidler, A.E.; Hopkins, G.A.; Wood, S.A. Geographically conserved microbiomes of four temperate water tunicates. Environ. Microbiol. Rep. 2016, 8, 470–478. [Google Scholar] [CrossRef]
- Dishaw, L.J.; Flores-Torres, J.; Lax, S.; Gemayel, K.; Leigh, B.; Melillo, D.; Mueller, M.G.; Natale, L.; Zucchetti, I.; De Santis, R.; et al. The gut of geographically disparate Ciona intestinalis harbors a core microbiota. PLoS ONE 2014, 9, e93386. [Google Scholar] [CrossRef]
- Evans, J.S.; Erwin, P.M.; Shenkar, N.; Lopez-Legentil, S. Introduced ascidians harbor highly diverse and host-specific symbiotic microbial assemblages. Sci. Rep. 2017, 7, 11033. [Google Scholar] [CrossRef] [Green Version]
- Dror, H.; Novak, L.; Evans, J.S.; Lopez-Legentil, S.; Shenkar, N. Core and dynamic microbial communities of two invasive ascidians: Can host-symbiont dynamics plasticity affect invasion capacity? Microb. Ecol. 2019, 78, 170–184. [Google Scholar] [CrossRef]
- Tianero, M.D.; Kwan, J.C.; Wyche, T.P.; Presson, A.P.; Koch, M.; Barrows, L.R.; Bugni, T.S.; Schmidt, E.W. Species specificity of symbiosis and secondary metabolism in ascidians. ISME J. 2015, 9, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Collado, L.; Inza, I.; Guarro, J.; Figueras, M.J. Presence of Arcobacter spp. in environmental waters correlates with high levels of fecal pollution. Environ. Microbiol. 2008, 10, 1635–1640. [Google Scholar] [CrossRef]
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, R.P. The gut microbiota of marine fish. Front. Microbiol. 2018, 9, 873. [Google Scholar] [CrossRef]
- Goddard-Dwyer, M.; Lopez-Legentil, S.; Erwin, P.M. Microbiome variability across the native and invasive range of the ascidian Clavelina oblonga. Appl. Environ. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Fernandez, N.; Beiras, R.; Vasconcelos, V. Toxicity assessment of crude and partially purified extracts of marine Synechocystis and Synechococcus cyanobacterial strains in marine invertebrates. Toxicon 2007, 50, 791–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, R.A.; Goeger, D.E.; Hills, P.; Mooberry, S.L.; Huang, N.; Romero, L.I.; Ortega-Barria, E.; Gerwick, W.H.; McPhail, K.L. Coibamide A, a potent antiproliferative cyclic depsipeptide from the Panamanian marine cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 2008, 130, 6324–6325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Engene, N.; Smith, J.E.; Preskitt, L.B.; Gerwick, W.H. Crossbyanols A−D, toxic brominated polyphenyl ethers from the Hawai’ian bloom-forming cyanobacterium Leptolyngbya crossbyana. J. Nat. Prod. 2010, 73, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Brinkhoff, T.; Bach, G.; Heidorn, T.; Liang, L.; Schlingloff, A.; Simon, M. Antibiotic production by a Roseobacter clade-affiliated species from the German Wadden Sea and its antagonistic effects on indigenous isolates. Appl. Environ. Microbiol. 2004, 70, 2560–2565. [Google Scholar] [CrossRef] [Green Version]
- Wiese, J.; Thiel, V.; Nagel, K.; Staufenberger, T.; Imhoff, J.F. Diversity of antibiotic-active bacteria associated with the brown alga Laminaria saccharina from the Baltic Sea. Mar. Biotechnol. 2009, 11, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Brilisauer, K.; Rapp, J.; Rath, P.; Schollhorn, A.; Bleul, L.; Weiss, E.; Stahl, M.; Grond, S.; Forchhammer, K. Cyanobacterial antimetabolite 7-deoxy-sedoheptulose blocks the shikimate pathway to inhibit the growth of prototrophic organisms. Nat. Commun. 2019, 10, 545. [Google Scholar] [CrossRef]
- Pujalte, M.J.; Lucena, T.; Ruvira, M.A.; Arahal, D.R.; Macián, M.C. The Family Rhodobacteraceae; Springer: Berlin/Heidelberg, Germany, 2014; pp. 439–512. [Google Scholar]
- Casso, M.; Turon, M.; Marco, N.; Pascual, M.; Turon, X. The microbiome of the worldwide invasive ascidian Didemnum vexillum. Front. Mar. Sci. 2020, 7, 201. [Google Scholar] [CrossRef]
- Rosenberg, E.; Zilber-Rosenberg, I. The hologenome concept of evolution after 10 years. Microbiome 2018, 6, 78. [Google Scholar] [CrossRef]
- da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, M.; Lindstrom, M.E.; Li, J. Fatty acid and lipid profiles with emphasis on n-3 fatty acids and phospholipids from Ciona intestinalis. Lipids 2015, 50, 1009–1027. [Google Scholar] [CrossRef] [PubMed]
- Menna, M. Important classes of bioactive alkaloids from marine ascidians: Structures, isolation and bioactivity. Curr. Top. Med. Chem. 2014, 14, 207–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauermeister, A.; Branco, P.C.; Furtado, L.C.; Jimenez, P.C.; Costa-Lotufo, L.V.; da Cruz Lotufo, T.M. Tunicates: A model organism to investigate the effects of associated-microbiota on the production of pharmaceuticals. Drug Discov. Today Dis. Models 2018, 28, 13–20. [Google Scholar] [CrossRef]
- Palanisamy, S.K.; Rajendran, N.M.; Marino, A. Natural products diversity of marine ascidians (Tunicates; Ascidiacea) and successful drugs in clinical development. Nat. Prod. Bioprospect. 2017, 7, 1–111. [Google Scholar] [CrossRef] [Green Version]
- Endo, H.; Hosoya, H.; Koyama, T.; Ichioka, M. Isolation of 10-hydroxypheophorbide a as a photosensitizing pigment from alcohol-treated Chlorella cells. Agric. Biol. Chem. 1982, 46, 2183–2193. [Google Scholar] [CrossRef]
- Schmidt, E.W.; Donia, M.S. Life in cellulose houses: Symbiotic bacterial biosynthesis of ascidian drugs and drug leads. Curr. Opin. Biotechnol. 2010, 21, 827–833. [Google Scholar] [CrossRef] [Green Version]
- Utermann, C.; Parrot, D.; Breusing, C.; Stuckas, H.; Staufenberger, T.; Blümel, M.; Labes, A.; Tasdemir, D. Combined genotyping, microbial diversity and metabolite profiling studies on farmed Mytilus spp. from Kiel Fjord. Sci. Rep. 2018, 8, 7983. [Google Scholar] [CrossRef]
- Erwin, P.M.; Carmen Pineda, M.; Webster, N.; Turon, X.; López-Legentil, S. Small core communities and high variability in bacteria associated with the introduced ascidian Styela plicata. Symbiosis 2012, 59, 35–46. [Google Scholar] [CrossRef]
- Johnson, C.H.; Patterson, A.D.; Idle, J.R.; Gonzalez, F.J. Xenobiotic metabolomics: Major impact on the metabolome. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 37–56. [Google Scholar] [CrossRef]
- Rochfort, S.J.; Ezernieks, V.; Maher, A.D.; Ingram, B.A.; Olsen, L. Mussel metabolomics—Species discrimination and provenance determination. Food Res. Int. 2013, 54, 1302–1312. [Google Scholar] [CrossRef]
- Wu, H.; Liu, X.; Zhang, X.; Ji, C.; Zhao, J.; Yu, J. Proteomic and metabolomic responses of clam Ruditapes philippinarum to arsenic exposure under different salinities. Aquat. Toxicol. 2013, 136–137, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Tanaka, A.; Nagai, K.; Suzuki, K.; Okada, G. New members of the trichothecene family. J. Antibiot. 1997, 50, 778–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbarskiĭ, V.B.; Lazhko, E.I.; Pomanova, N.P.; Fomicheva, E.V.; Saburova, T.P. Rubomycins M and N-new anthracycline antibiotics. Russ. J. Bioorg. Chem. 1991, 17, 1698–1701. [Google Scholar]
- Macel, M.; de Vos, R.C.; Jansen, J.J.; van der Putten, W.H.; van Dam, N.M. Novel chemistry of invasive plants: Exotic species have more unique metabolomic profiles than native congeners. Ecol. Evol. 2014, 4, 2777–2786. [Google Scholar] [CrossRef] [PubMed]
- Wolf, V.C.; Berger, U.; Gassmann, A.; Müller, C. High chemical diversity of a plant species is accompanied by increased chemical defence in invasive populations. Biol. Invasions 2011, 13, 2091–2102. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OTU | Lowest Classification (BLAST) | Accession Number (BLAST) | Isolation Source According To BLAST | Dominant Sample Type (Location) This Study |
|---|---|---|---|---|
| OTU1 | Kordiimonas sp. | KF494349.1 | Ciona intestinalis (tunic) | Tunic |
| OTU2 | Rhizobiales | MN006421.1 | Various, e.g., Ciona intestinalis (tunic) | Tunic |
| OTU3 | Arenibacter sp. | KF494352.1 | Ciona intestinalis (tunic) | Tunic |
| OTU4 | Pseudomonas sp. | MH244157.1 | Sediment | Gut (Kiel) |
| OTU6 | Synechococcus sp. | MH358353.1 | Marine environment | Gut |
| OTU7 | Uncultured alphaproteobacterium_1-21 | FJ659126.1 | Ascidian (Aplidium conicum; tunic) | Tunic |
| OTU8 | Synechococcus sp. | KU867940.1 | Seawater | Gut |
| OTU10 | Ubc Woods-Hole_a5143 | KF798938.1 | Ciona intestinalis (gut) | Gut |
| OTU11 | Rhodobacteraceae | KU173743.1 | Seawater | Seawater |
| OTU13 | Bifidobacterium dentium | LR134349.1 | Human | Gut (Helgoland) |
| OTU15 | Roseobacter sp. | MK224709.1 | Red algae | Gut (Canada) |
| OTU16 | Ubc Woods-Hole_a5449 | KF799049.1 | Ciona intestinalis (gut) | Gut |
| OTU17 | Litoreibacter sp. | KJ513684.1 | Seawater | Several |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Utermann, C.; Blümel, M.; Busch, K.; Buedenbender, L.; Lin, Y.; Haltli, B.A.; Kerr, R.G.; Briski, E.; Hentschel, U.; Tasdemir, D. Comparative Microbiome and Metabolome Analyses of the Marine Tunicate Ciona intestinalis from Native and Invaded Habitats. Microorganisms 2020, 8, 2022. https://doi.org/10.3390/microorganisms8122022
Utermann C, Blümel M, Busch K, Buedenbender L, Lin Y, Haltli BA, Kerr RG, Briski E, Hentschel U, Tasdemir D. Comparative Microbiome and Metabolome Analyses of the Marine Tunicate Ciona intestinalis from Native and Invaded Habitats. Microorganisms. 2020; 8(12):2022. https://doi.org/10.3390/microorganisms8122022
Chicago/Turabian StyleUtermann, Caroline, Martina Blümel, Kathrin Busch, Larissa Buedenbender, Yaping Lin, Bradley A. Haltli, Russell G. Kerr, Elizabeta Briski, Ute Hentschel, and Deniz Tasdemir. 2020. "Comparative Microbiome and Metabolome Analyses of the Marine Tunicate Ciona intestinalis from Native and Invaded Habitats" Microorganisms 8, no. 12: 2022. https://doi.org/10.3390/microorganisms8122022