
Molecular Networking-Guided Isolation of New Etzionin-Type Diketopiperazine Hydroxamates from the Persian Gulf Sponge Cliona celata

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Bioactivity Profiling of the Crude Sponge Extract
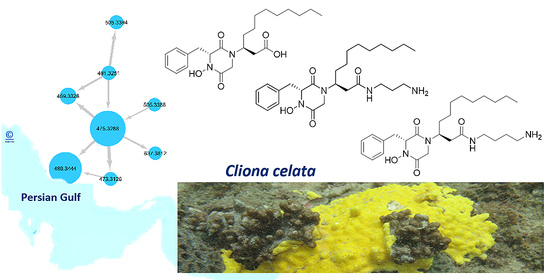
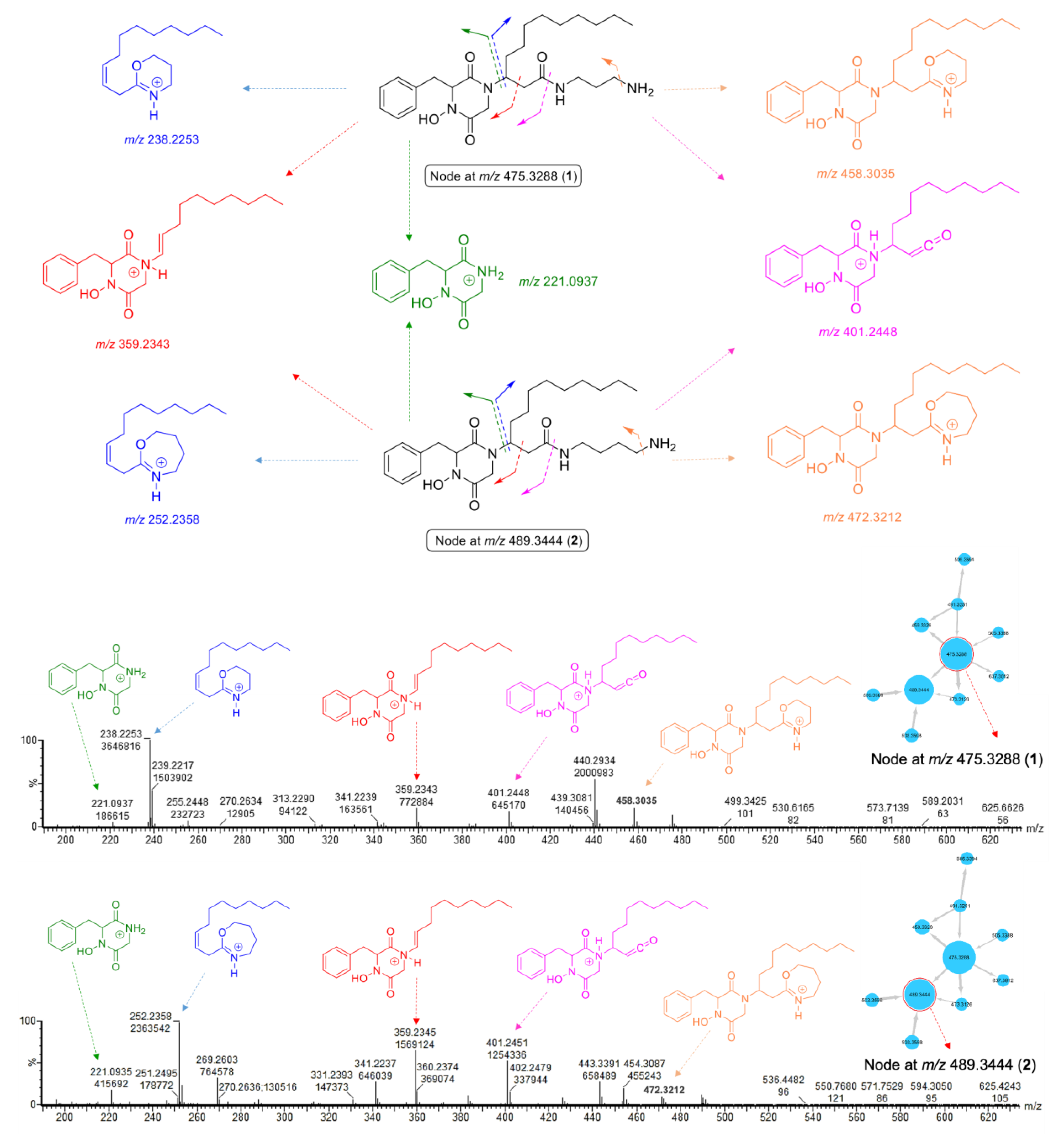
2.2. MS/MS Molecular-Networking-Based Dereplication of the Crude Extract
2.3. Purification and Structure Elucidation of DKPHs
2.4. Bioactivity of Compounds 1 and 2
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Sponge Material
4.3. Extraction and Isolation
4.4. UPLC-QToF-MS/MS Analyses
4.5. Molecular Networking
4.6. Antimicrobial Activity
4.7. Cytotoxic Activity
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Erpenbeck, D.; Gholami, A.; Hesni, M.A.; Ranjbar, M.S.; Galitz, A.; Eickhoff, B.; Namuth, L.; Schumacher, T.; Esmaeili, H.R.; Wörheide, G. Molecular biodiversity of Iranian shallow water sponges. Syst. Biodiver. 2020, 18, 192–202. [Google Scholar] [CrossRef]
- Sheppard, C.R. Physical environment of the Gulf relevant to marine pollution: An overview. Mar. Pollut. Bull. 1993, 27, 3–8. [Google Scholar] [CrossRef]
- Braulik, G.T.; Ranjbar, S.; Owfi, F.; Aminrad, T.; Dakhteh, S.M.H.; Kamrani, E.; Mohsenizadeh, F. Marine mammal records from Iran. J. Cetacean Res. Manag. 2010, 11, 49–63. [Google Scholar]
- Soest, R.W.M.V.; Esnault, N.B.; Hooper, J.N.A.; Rützler, K.; Voogd, N.J.D.; Alvarez, B.; Hajdu, E.; Pisera, A.B.; Manconi, R.; Schönberg, C.; et al. “Cliona Celata Grant, 1826”, World Porifera Database. 26 February 2010. Available online: http://marinespecies.org/porifera/porifera.php?p=taxdetails&id=134121 and http://www.marinespecies.org (accessed on 21 December 2020).
- Carballo, J.; Moyano, J.S.; Gómez, J.G. Taxonomic and ecological remarks on boring sponges (Clionidae) from the Straits of Gibraltar (southern Spain): Tentative bioindicators? Zool. J. Linnean. Soc. 1994, 112, 407–424. [Google Scholar] [CrossRef]
- Custódio, M.R.; Symposium, R.I.S. Porifera Research: Biodiversity, Innovation and Sustainability; Museu Nacional: Rio de Janeiro, Brazil, 2007; pp. 1–694. [Google Scholar]
- Bary, K.; Elamraoui, B.; Bamhaoud, T. Chemical characterization of Cliona viridis: Sponge of Atlantic Moroccan Coast. Int. J. Sci. 1997, 26, 14–22. [Google Scholar]
- Erdman, T.; Thomson, R. Sterols from the sponges Cliona celata Grant and Hymeniacidon perleve Montagu. Tetrahedron 1972, 28, 5163–5173. [Google Scholar] [CrossRef]
- Lenis, L.A.; Nuñez, L.; Jiménez, C.; Riguera, R. Isonitenin and acetylhomoagmatine new metabolites from the sponges Spongia officinalis and Cliona celata collected at the Galician coast (NW Spain). J. Nat. Prod. 1996, 8, 15–23. [Google Scholar]
- Sawangwong, P.; Wattanadilok, R.; Kijjoa, A.; Silva, A.M.; Eaton, G.; Herz, W. Secondary metabolites from a marine sponge Cliona patera. Biochem. Syst. Ecol. 2008, 5, 493–496. [Google Scholar] [CrossRef]
- Andersen, R.J.; Stonard, R.J. Clionamide, a major metabolite of the sponge Cliona celata Grant. Can. J. Chem. 1979, 57, 2325–2328. [Google Scholar] [CrossRef] [Green Version]
- Stonard, R.J.; Andersen, R.J. Celenamides A and B, linear peptide alkaloids from the sponge Cliona celata. J. Org. Chem. 1980, 45, 3687–3691. [Google Scholar] [CrossRef]
- Palermo, J.A.; Rodríguez Brasco, M.F.; Cabezas, E.; Balzaretti, V.; Seldes, A.M. Celenamide E, a tripeptide alkaloid from the Patagonian sponge Cliona chilensis. J. Nat. Prod. 1998, 61, 488–490. [Google Scholar] [CrossRef]
- Stonard, R.J.; Andersen, R.J. Linear peptide alkaloids from the sponge Cliona celata (Grant). Celenamides C and D. Can. J. Chem. 1980, 58, 2121–2126. [Google Scholar] [CrossRef]
- Nazemi, M.; Gilkolai, F.R.; Lakzaei, F.; Pishvarzad, F.; Ahmadzadeh, O. First record on the distribution and abundance of three sponge species from Hormoz island, Persian Gulf-Iran. BFAIJ 2015, 7, 72–78. [Google Scholar]
- Kouchaksaraee, R.M.; Farimani, M.M.; Li, F.; Nazemi, M.; Tasdemir, D. Integrating molecular networking and 1H NMR spectroscopy for isolation of bioactive metabolites from the Persian Gulf sponge Axinella sinoxea. Mar. Drugs 2020, 18, 366. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, A.; Villar-Briones, M.; Orešiˇc, M. MZmine2: Modular framework for processing, visualizing and analyzing mass spectrometry-based molecular profile data. BMC Bioinf. 2010, 11, 395. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kossuga, M.H.; Lira, S.P.; McHugh, S.; Torres, Y.R.; Lima, B.A.; Gonçalves, R.; Veloso, K.; Ferreira, A.G.; Rocha, R.M.; Berlinck, R.G. Antibacterial modified diketopiperazines from two ascidians of the genus Didemnum. J. Braz. Chem. Soc. 2009, 20, 704–711. [Google Scholar] [CrossRef]
- Hirsch, S.; Miroz, A.; McCarthy, P.; Kashman, Y. Etzionin, a new antifungal metabolite from a red sea tunicate. Tetrahedron Lett. 1989, 30, 4291–4294. [Google Scholar] [CrossRef]
- Bystrov, V.F. Spin-spin coupling and the conformational states of peptide systems. Prog. Nucl. Magn. Reson. Spectrosc. 1976, 10, 41–82. [Google Scholar] [CrossRef]
- Moneda, J.D.; Leal, K.Z.; Seidl, P.R.; Pimentel, H.R.X. Structural dependence of long-range carbon-hydrogen coupling in camphor. Ann. Magn. Reson. 2003, 2, 41–82. [Google Scholar]
- Vaz, E.; Suarez, M.F.; Muñoz, L. Determination of the absolute stereochemistry of etzionin. Tetrahedron Asymmetry 2003, 14, 1935–1942. [Google Scholar] [CrossRef]
- Fonnegra, C.A.; Zea, S. Observations on reef coral undermining by the Caribbean excavating sponge Cliona delitrix (Demospongiae, Hadromerida). In Porifera Research: Biodiversity, Innovation and Sustainability; Custódio, M.R., Hajdu, G.L., Hajdu, E., Muricy, Eds.; Museu Nacional: Rio de Janeiro, Brazil, 2007; Volume 28, pp. 247–254. [Google Scholar]
- Cerrano, C.; Calcinai, B.; Di Camillo, C.G.; Valisano, L.; Bavestrello, G. How and why do sponges incorporate foreign material? Strategies in Porifera. In Porifera Research: Biodiversity, Innovation and Sustainability; Custódio, M.R., Hajdu, G.L., Hajdu, E., Muricy, G., Eds.; Museu Nacional: Rio de Janeiro, Brazil, 2007; Volume 28, pp. 239–246. [Google Scholar]
- Teragawa, C.K. Particle transport and incorporation during skeleton formation in a keratose sponge: Dysidea etheria. Biol. Bull. 1986, 170, 321–334. [Google Scholar] [CrossRef]
- Snowden, E. Cliona celata A sponge. In Marine Life Information Network: Biology and Sensitivity Key Information Reviews; Tyler-Walters, H., Hiscock, K., Eds.; Marine Biological Association of the United Kingdom: Plymouth, UK, 2007; Available online: https://www.marlin.ac.uk/species/detail/2188 (accessed on 29 April 2021).
- Fonnegra, A.C.; Victoria, M.L.; Velandia, F.P.; Zea, S. Ecología química de las esponjas excavadoras Cliona aprica, C. caribbaea, C. delitrix y C. tenuis. Bol. Investig. Mar. Cost. 2005, 34. [Google Scholar] [CrossRef]
- Fonnegra, A.C.; Castellanos, L.; Zea, S.; Duque, C.; Rodríguez, J.; Jiménez, C. Clionapyrrolidine A—A metabolite from the encrusting and excavating sponge Cliona tenuis that kills coral tissue upon contact. J. Chem. Ecol. 2008, 34, 1565–1574. [Google Scholar] [CrossRef]
- Fattorusso, E.; Scafati, O.T.; Petrucci, F.; Bavestrello, G.; Calcinai, B.; Cerrano, C.; Di Meglio, P.; Ianaro, A. Polychlorinated androstanes from the burrowing sponge Cliona nigricans. Org. Lett. 2004, 6, 1633–1635. [Google Scholar] [CrossRef]
- Keyzers, R.A.; Daoust, J.; Coleman, M.T.D.; Soest, R.V.; Balgi, A.; Donohue, E.; Roberge, M.; Andersen, R.J. Autophagy-modulating aminosteroids isolated from the sponge Cliona celata. Org. Lett. 2008, 10, 2959–2962. [Google Scholar] [CrossRef]
- Andersen, R.J. Tetracetyl clionamide, a 6-bromotryptophan derivative from the sponge Cliona celata. Tetrahedron Lett. 1978, 19, 2541–2544. [Google Scholar] [CrossRef]
- Palermo, J.A.; Brasco, M.F.R.; Seldes, A.M. Storniamides A-D: Alkaloids from a Patagonian sponge Cliona sp. Tetrahedron 1996, 52, 2727–2734. [Google Scholar] [CrossRef]
- Borthwick, A.D. 2, 5-Diketopiperazines: Synthesis, reactions, medicinal chemistry, and bioactive natural products. Chem. Rev. 2012, 112, 3641–3716. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.M.; Yi, X.X.; Zhou, Y.; Su, X.; Peng, Y.; Gao, C.H. An update on 2, 5-diketopiperazines from marine organisms. Mar. Drugs 2014, 12, 6213–6235. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.R.; Santos, M.F.; Williams, D.E.; Andersen, R.J.; Padula, V.; Ferreira, A.G.; Berlinck, R.G. Rodriguesic acids, modified diketopiperazines from the gastropod mollusc Pleurobranchus areolatus. J. Braz. Chem. Soc. 2014, 25, 788–794. [Google Scholar] [CrossRef]
- Vergne, C.; Esnault, N.B.; Perez, T.; Martin, M.T.; Adeline, M.T.; Dau, E.T.H.; Al-Mourabit, A. Verpacamides A-D, a sequence of C11N5 diketopiperazines relating Cyclo (Pro-Pro) to Cyclo (Pro-Arg), from the marine sponge Axinella vaceleti: Possible biogenetic precursors of pyrrole-2-aminoimidazole alkaloids. Org. Lett. 2006, 8, 2421–2424. [Google Scholar] [CrossRef] [PubMed]
- Fenical, W.; Jensen, P.R.; Cheng, X.C. Halimide, A Cytotoxic Marine Natural Product, and Derivatives Thereof. U.S. Patent US6358957B1, 30 May 2000. [Google Scholar]
- Cimino, P.J.; Huang, L.; Du, L.; Wu, Y.; Bishop, J.; Dalsing-Hernandez, J.; Kotlarczyk, K.; Gonzales, P.; Carew, J.; Nawrocki, S. Plinabulin, an inhibitor of tubulin polymerization, targets KRAS signaling through disruption of endosomal recycling. Biomed. Rep. 2019, 10, 218–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, B.; Hooper, J.N. Taxonomic revision of the order Halichondrida (Porifera: Demospongiae) of northern Australia: Family Axinellidae. Int. J. Soc. 2009, 25, 17–42. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2019, 13, 426. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH, Mult. (J in Hz) | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| 1 (N) | - | - | - |
| 2 | - | - | - |
| 3 | 4.48 (m) | 4.42 (m) | 4.50 (m) |
| 4 (N) | - | - | - |
| 5 | - | - | - |
| 6 | 3.47 (d 16.8) 2.51 (d 16.8) | 3.47 (d 16.6) 2.60 (d 16.6) | 3.52 (d 17.3) 2.41 (d 17.3) |
| 7 | 3.18 (m) 3.45 (m) | 3.15 (dd 13.8, 2.7) 3.47 (m) | 3.18 (dd 13.8, 2.7) 3.43 (m) |
| 8 | - | - | - |
| 9 | 7.20 (d 7.1) | 7.20 (d 7.1) | 7.19 (m) |
| 10 | 7.30 (m) | 7.28 (m) | 7.30 (m) |
| 11 | 7.30 (m) | 7.28 (m) | 7.31 (m) |
| 12 | 7.30 (m) | 7.28 (m) | 7.30 (m) |
| 13 | 7.20 (m) | 7.20 (m) | 7.19 (m) |
| 1′ | - | - | - |
| 2′ | 2.16 (dd 14.2, 7.7) 2.41 (m) | 2.16 (dd 14.2, 6.6) 2.60 (m) | 2.15 (dd 15.16, 7.23) 2.27 (m) |
| 3′ | 4.24 (br) | 3.93 (br) | 4.57 (m) |
| 4′ | 1.51 (m) 1.57 (m) | 1.53 (m) 1.63 (m) | 1.48 (m) 1.57 (m) |
| 5′ | 1.08 (m) | 1.07 (m) | 1.10 (m) |
| 6–′9′ | ~1.27 (m) | ~1.29 (m) | ~1.27 (m) |
| 10′ | 1.28 (m) | 1.29 (m) | 1.28 (m) |
| 11′ | 1.32 (m) | 1.32 (m) | 1.32 (m) |
| 12′ | 0.91 (t 7.1) | 0.91 (t 7.1) | 0.90 (t 7.1) |
| 1″ | 3.17 (m) 3.32 (m) | 3.02 (m) 3.37 (m) | - |
| 2″ | 1.77 (p 7.2) | 1.54 (m) | - |
| 3″ | 2.85 (t 7.2) | 1.55 (m) | - |
| 4″ | - | 2.87 (m) | - |
| Position | δC, Type | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| 1 (N) | - | - | - |
| 2 | 167.3 (C) | 167.3 (C) | 166.9 (C) |
| 3 | 66.7 (CH) | 67.0 (CH) | 66.3 (CH) |
| 4 (N) | - | - | - |
| 5 | 162.2 (C) | 161.3 (C) | 163.1 (C) |
| 6 | 47.4 (CH2) | n.o. 1 | 45.7 (CH2) |
| 7 | 36.4 (CH2) | 36.4 (CH2) | 36.4 (CH2) |
| 8 | 136.1 (C) | 136.3 (C) | 135.8 (C) |
| 9 | 131.5 (CH) | 131.5 (CH) | 131.4 (CH) |
| 10 | 129.7 (CH) | 129.7 (CH) | 129.8 (CH) |
| 11 | 128.7 (CH) | 128.7 (CH) | 128.8 (CH) |
| 12 | 129.7 (CH) | 129.7 (CH) | 129.8 (CH) |
| 13 | 131.5 (CH) | 131.5 (CH) | 131.4 (CH) |
| 1′ | 173.2 (C) | 172.9 (C) | 175.8 (C) |
| 2′ | 39.1 (CH2) | 39.2 (CH2) | 38.9 (CH2) |
| 3′ | 58.1 (CH) | n.o. 1 | n.o. 1 |
| 4′ | 31.4 (CH2) | 31.6 (CH2) | 31.4 (CH2) |
| 5′ | 27.2 (CH2) | 27.4 (CH2) | 27.1 (CH2) |
| 6′–9′ | 30.2-30.06 (CH2) | 30.2-30.06 (CH2) | 30.2-30.06 (CH2) |
| 10′ | 33.0 (CH2) | 33.0 (CH2) | 33.0 (CH2) |
| 11′ | 23.7 (CH2) | 23.7 (CH2) | 23.7 (CH2) |
| 12′ | 14.4 (CH3) | 14.4 (CH3) | 14.4 (CH3) |
| 1″ | 37.1 (CH2) | 39.7 (CH2) | - |
| 2″ | 29.8 (CH2) | 27.7 (CH2) | - |
| 3″ | 38.6 (CH2) | 26.5 (CH2) | - |
| 4″ | - | 40.4 (CH2) | - |
| Sample | IC50 Values (in µM) | |||||||
|---|---|---|---|---|---|---|---|---|
| E. faecium | MRSA | E. coli | C. albicans | C. neoformans | A375 | HCT116 | HaCaT | |
| Compound 1 | 19.6 | 46.2 | 159.1 | 116.0 | 22.6 | 43.2 | 59.9 | 50.0 |
| Compound 2 | 36.3 | 44.7 | 152.9 | 125.0 | 28.1 | 37.9 | 39.5 | 37.5 |
| Positive control | 0.57 | 4.6 | 4.7 | 2.3 | 0.2 | 0.2 | 0.4 | 42.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouchaksaraee, R.M.; Li, F.; Nazemi, M.; Farimani, M.M.; Tasdemir, D. Molecular Networking-Guided Isolation of New Etzionin-Type Diketopiperazine Hydroxamates from the Persian Gulf Sponge Cliona celata. Mar. Drugs 2021, 19, 439. https://doi.org/10.3390/md19080439
Kouchaksaraee RM, Li F, Nazemi M, Farimani MM, Tasdemir D. Molecular Networking-Guided Isolation of New Etzionin-Type Diketopiperazine Hydroxamates from the Persian Gulf Sponge Cliona celata. Marine Drugs. 2021; 19(8):439. https://doi.org/10.3390/md19080439
Chicago/Turabian StyleKouchaksaraee, Reza Mohsenian, Fengjie Li, Melika Nazemi, Mahdi Moridi Farimani, and Deniz Tasdemir. 2021. "Molecular Networking-Guided Isolation of New Etzionin-Type Diketopiperazine Hydroxamates from the Persian Gulf Sponge Cliona celata" Marine Drugs 19, no. 8: 439. https://doi.org/10.3390/md19080439