Coastal upwelling systems as dynamic mosaics of bacterioplankton functional specialization
 Erick Delgadillo-Nuño1
Erick Delgadillo-Nuño1  Eva Teira1
Eva Teira1  Benjamin Pontiller2,3
Benjamin Pontiller2,3  Daniel Lundin2 Vanessa Joglar1
Daniel Lundin2 Vanessa Joglar1  Carlos Pedrós-Alió4
Carlos Pedrós-Alió4  Emilio Fernández1
Emilio Fernández1  Jarone Pinhassi2
Jarone Pinhassi2  Sandra Martínez-García1*
Sandra Martínez-García1*- 1Departamento de Ecoloxía e Bioloxía Animal, Centro de Investigación Mariña da Universidade de Vigo (CIM-UVigo), Vigo, Spain
- 2Centre for Ecology and Evolution in Microbial Model Systems, Linnaeus University, Kalmar, Sweden
- 3GEOMAR Helmholtz Centre for Ocean Research Kiel, Kiel, Germany
- 4Centro Nacional de Biotecnología (CNB-CSIC), Madrid, Spain
Coastal upwelling areas are extraordinarily productive environments where prokaryotic communities, the principal remineralizers of dissolved organic matter (DOM), rapidly respond to phytoplankton bloom and decay dynamics. Nevertheless, the extent of variability of key microbial functions in such dynamic waters remains largely unconstrained. Our metatranscriptomics analyses of 162 marker genes encoding ecologically relevant prokaryotic functions showed distinct spatial-temporal patterns in the NW Iberian Peninsula upwelling area. Short-term (daily) changes in specific bacterial functions associated with changes in biotic and abiotic factors were superimposed on seasonal variability. Taxonomic and functional specialization of prokaryotic communities, based mostly on different resource acquisition strategies, was observed. Our results uncovered the potential influence of prokaryotic functioning on phytoplankton bloom composition and development (e.g., Cellvibrionales and Flavobacteriales increased relative gene expression related to vitamin B12 and siderophore metabolisms during Chaetoceros and Dinophyceae summer blooms). Notably, bacterial adjustments to C- or N-limitation and DMSP availability during summer phytoplankton blooms and different spatial-temporal patterns of variability in the expression of genes with different phosphate affinity indicated a complex role of resource availability in structuring bacterial communities in this upwelling system. Also, a crucial role of Cellvibrionales in the degradation of DOM (carbohydrate metabolism, TCA cycle, proteorhodopsin, ammonium, and phosphate uptake genes) during the summer phytoplankton bloom was found. Overall, this dataset revealed an intertwined mosaic of microbial interactions and nutrient utilization patterns along a spatial-temporal gradient that needs to be considered if we aim to understand the biogeochemical processes in some of the most productive ecosystems in the world´s oceans.
Introduction
Coastal upwelling areas are complex and dynamic environments where winds displace surface water off the coast, allowing nutrient-rich deep water to reach the surface and driving remarkable productivity at all trophic levels (ultimately sustaining major portions of global fisheries) (Kämpf and Chapman, 2016; Stock et al., 2017) [see Kämpf, J. and Chapman, P 2016 and references therein]. Bacterioplankton is a crucial component of the ocean biome that has long been recognized for its principal role in regulating the biogeochemical cycling of carbon (Rivkin and Legendre, 2001), but also the remineralization and utilization of elements like N and P that are vital for ocean productivity (Caron, 1994). Moreover, recent findings emphasize the role of bacteria and their interactions with phytoplankton in providing vitamins and growth factors necessary for organisms throughout the food web (Shilova et al., 2014; Amin et al., 2015). Still, the specific adjustments in bacterial metabolism that regulate such processes in upwelling areas, and the taxa involved, remain largely unexplored.
The usage of ‘meta-omics’ techniques has allowed the scientific community to gain comprehensive understanding on the succession of specific prokaryotic taxa and their metabolisms operating during the evolution of phytoplanton blooms. Thus, a recent study based on mesocosm experiments carried out in summer in the NW Iberian Peninsula upwelling system, showed important daily changes in the transcription of glycoside hydrolases, peptidases, and transporters, supporting the hypothesis that resource partitioning is affected by temporal changes in the availability of dissolved organic matter (DOM) (Pontiller et al., 2022). Similarly, recurrent patterns at the functional level, particularly regarding substrate-induced responses, have been observed throughout bacterioplankton succession during phytoplankton blooms in the southern North Sea (Teeling et al., 2012; Teeling et al., 2016) and in the Monterey upwelling system (Nowinski and Moran, 2021). Although geographically distant, there are interesting similarities in the involvement of bacterial taxa in these studies, with pronounced contributions of Flavobacteriaceae (Bacteroidota; recognized for their polymer degrading capacities) along with Alphaproteobacteria (e.g. Roseobacter) and Gammaproteobacteria (e.g. Alteromonadales and Cellvibrionales) that differ in preference for different quantity and quality of resources. Nevertheless, and surprisingly, little is known about the temporal shifts in the functional profiles of different bacteria associated with changes in nutrient availability, hydrodynamic conditions, or phytoplankton community composition and activity along the coast to offshore spatial gradient in upwelling systems.
The environmental conditions for life in upwelling areas vary substantially along both spatial and temporal dimensions (Kämpf and Chapman, 2016). A key axis in the spatial structuring of upwelling ecosystems is the transition from the coastal zone, where the upwelling occurs, to offshore areas, where surface waters are displaced over time. This axis is strongly influenced by different coastal features like interactions between winds and the coastal topography (Figueiras et al., 2002), potential riverine inputs (Teixeira et al., 2018), and tidal forcing (Souto et al., 2001), generating spatial heterogeneity at various scales. In this regard, Satinsky et al. (2017) suggested that changes in microbial element cycling along the coastal system of the Amazon River Plume are largely associated with shifts in specific metabolisms. Accordingly, important spatial and temporal variability in the structure and function of microbial communities in large and heterogeneous upwelling areas can be expected (Zdanowski and Figueiras, 1997; Arístegui et al., 2009; Cury et al., 2011; Bertrand and Allen, 2012; Joglar et al., 2021a).
Short-term variability in wind regimes superimposed on seasonality induces patchiness in the prevalence of upwelling and downwelling events (typically 2-5 days (Pisareva et al., 2019)), particularly in the coastal zone (Blanton et al., 1984). Measurements of microbial bulk abundance and activity (e.g., production, respiration, enzymatic activities) show that such short-term temporal dynamics are associated with phytoplankton blooms and affect microbial community biomass, activity, and composition (Souto et al., 2001). The spatial and temporal scales of variability of phytoplankton growth and microbial food web functioning in upwelling systems are, therefore, inherently complex (Kerkhof et al., 1999; Barbosa et al., 2001; Bergen et al., 2015; Wear et al., 2015; Hernández-Ruiz et al., 2018), and constraining this complexity remains an important challenge.
The coastal area of the NW Iberian Peninsula is a highly dynamic and productive ecosystem, characterized by a light–limited winter and a nutrient–limited summer period, and by diatom and dinoflagellate blooms during spring and autumn, respectively (Figueiras and Ríos, 1993). On top of these seasonal patterns, this region is affected by the intermittent upwelling of cold and inorganic nutrient-rich Eastern North Atlantic Central Water (Nogueira et al., 1997). Although upwelling-favorable northerly winds prevail from March to September and downwelling-favorable southerly winds the rest of the year, out-of-season upwelling or downwelling events have been frequently recorded. During downwelling periods, surface, nutrient-poor seawater moves from the ocean to the coast. The extensive knowledge of the physical, chemical, and biological oceanography in the area provides a solid background against which to assess aspects of the microbial ecology (Souto et al., 2001; Figueiras et al., 2002; Teira et al., 2015; Teixeira et al., 2018; Joglar et al., 2021a). Thus, we approached this ecosystem with the objective of refining our understanding of microbial and biogeochemical processes in upwelling areas. In the current work we aimed to capture the range of variability in frequencies and amplitudes of key microbial functions. Our comparative metatranscriptome analysis comprised a collection of 162 genes previously used as informative markers for nutrient (e.g., C, N, P, S, Fe) fluxes and microbial metabolism (e.g., response to stimuli and stress, phototrophy, and synthesis of vitamins) (Satinsky et al., 2014; Robidart et al., 2019) and covered different oceanographic and biological settings. We hypothesized that short-term oceanographic features as wells as differences in upwelling intensity between coastal and offshore areas will have distinct effects on the diverse set of key microbial functions studied.
Methods
Study site and sampling
Three 10-day cruises were conducted on board B/O Ramón Margalef in February (winter), April (spring), and August (summer) 2016 to cover a wide range of hydrographic and ecological conditions. Samples were taken at two locations in the eastern Atlantic Ocean, in the upwelling system near the Ría de Vigo: the Coastal station (88 m depth, 42.14° N, 8.95° W, sampled between 8:00 and 8:20 am) and the Offshore station (260 m depth, 42.10° N, 9.60° W, sample between 6:30 and 6:45pm) (Figure 1A). It is important to note that the different time of sampling may have biased the metatranscriptome comparisons between both sampling stations, as it has been previously shown that prokaryotic transcriptomes from natural samples may exhibit diel periodicity, particularly those of Cyanobacteria in open ocean waters (Ottesen et al., 2013; Ottesen et al., 2014).
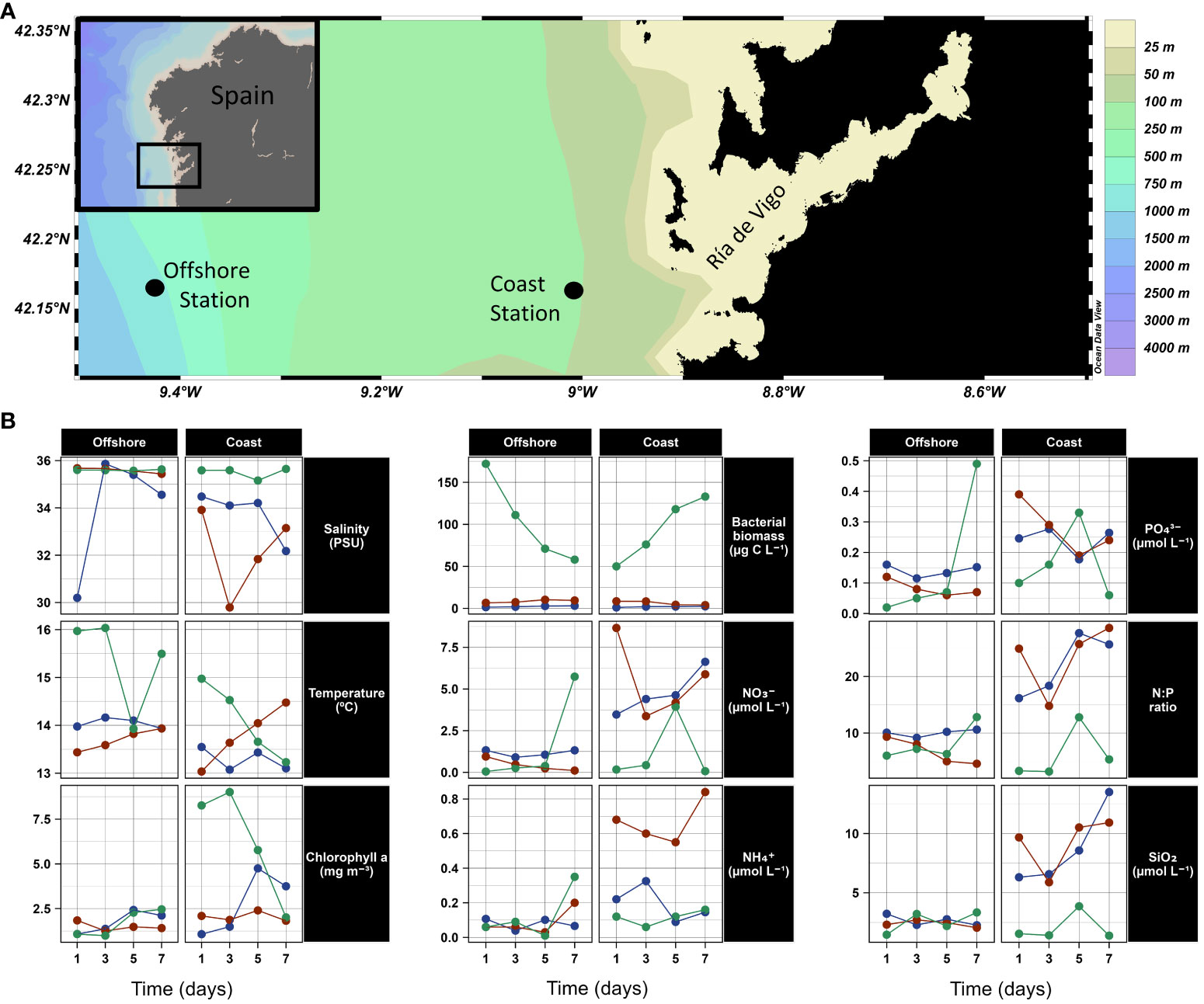
Figure 1 (A) Map showing bathymetry and sampling locations (coastal and offshore) in NW Iberian Peninsula. (B) Temporal evolution of salinity, temperature, chlorophyll a, bacterial abundance, nitrate (NO3-), ammonium (NH₄⁺), phosphate (PO₄³⁻), N:P ratio (calculated as the sum of NO3-, NO2- and NH₄⁺divided by PO₄³⁻), and silicate (SiO2) at surface (5m) during winter (blue), spring (red) and summer (green) at surface (5m) at days 1, 3, 5 and 7 at the offshore and coastal stations.
Detailed information on the experimental setup and specific methods below are given in Supplementary Material.
Hydrographic survey
Vertical profiles of temperature (°C), salinity, turbidity (NTU), total chlorophyll fluorescence, and photosynthetically active radiation (PAR) were obtained using a Seabird CTD rosette. Inorganic-nutrient determinations of collected water were done with a Bran + Luebbe segmented flow analyzer (Hansen and Grasshoff, 1983).
Chlorophyll a and prokaryotic abundance
Chlorophyll a (Chl-a) concentration was measured by the non-acidification technique as a phytoplankton biomass proxy (Welschmeyer, 1994). Fluorescence was determined with a TD-700 Turner Designs fluorometer (absorption coefficient as 87.7 at 663 nm) (Lorenzen and Newton Downs, 1986). Prokaryotic abundance in seawater samples was calculated as described by Gasol and Del Giorgio (2000) using a Becton Dickinson FACSCalibur flow cytometer (488 nm light produced by argon‐ion laser).
Microbial community composition
Community composition was assessed from 24 samples by sequencing the 16S rRNA gene for prokaryotes and the 18S rRNA gene for eukaryotes (0.22 - 3µm size-fraction and > 3µm size-fraction, respectively). DNA was amplified using the universal primers 515F and 926R for prokaryotes (Parada et al., 2016) and TAReuk454FWD1 and TAReukREV3 for eukaryotes (Logares et al., 2014). Amplified regions were sequenced in an Illumina Miseq platform. The sequencing depth for amplicon 16S and 18S was 10,000 reads per sample The sequences obtained were analyzed with the software DADA2 for amplicon sequence variants (ASVs) (Callahan et al., 2016) using the SILVA reference database (v138.1) for taxonomic assignment of 16S (Quast et al., 2013), and the databases PR2 (Guillou et al., 2012) and the marine protist from the BioMarKs project (Massana et al., 2015) for taxonomic assignment of 18S ASVs.
Metatranscriptomic analyses: prokaryotic community gene expression
RNA samples (< 3µm size-fraction) were taken during the experimental period at the surface in the coastal and oceanic stations (a total of 24 samples). Sampling for gene expression analyses was carried out on days 1, 3, 5, and 7 of each cruise. mRNA samples were extracted and sequenced using Illumina Miseq sequencing technology at the Science for Life Laboratory in Stockholm (SciLifeLab; www.scilifelab.se). The sequencing depth was 70,000 sequences. Illumina HiSeq 2500 raw paired-end reads (2x125bp) were quality checked with FastQC and MultiQC (Ewels et al., 2016), primer sequences were removed with cutadapt (Marcel, 2011), reads were trimmed with Sickle (Joshi and Fass, 2011) and filtered with ERNE (Del Fabbro et al., 2013) against a “contamination reference” database to filter reads designated as artifacts. Subsequently, forward and reverse reads were merged with PEAR (Zhang et al., 2014). All Merged reads were aligned with DIAMOND (Buchfink et al., 2014) against the NCBI RefSeq protein database (O’Leary et al., 2016). Afterward, all the sequences were compared to the 162 protein families (Pfams and TIGRfams) by running MEGAN (Huson et al., 2016) to annotate taxonomy and all possible transcripts to select marker genes relevant to a variety of ecological processes. The 162 genes found out of the 198 target protein profiles were grouped into nine categories and 44 subcategories (Table S1). To make comparisons between samples, considering that the transcripts have different lengths, and the libraries differ in size, we used a Transcript Per Million (TPM) normalization.
Statistical analyses
Non-parametric Mann-Whitney U Test analyses were performed to compare the salinity, nutrient concentrations, bacterial abundance and chlorophyll a concentration during the different seasons at both stations. T-tests analyses were performed to compare the contribution of each taxonomical group to the 16S dataset and to the metatranscriptome. To test the null hypothesis that data came from a normally distributed population, a Kolmogorov-Smirnov & Lillierors test was used.
Analysis of Non-parametric MultiDimensional Scaling (nMDS) and PERmutational Multivariate ANalysis Of VAriance (PERMANOVA) were used to test the grouping of metatranscriptomes by seasons and stations. ReDundancy Analyses (RDA) were applied to show the variability in the metatranscriptomes that is explained by environmental data and by the composition of phytoplankton and bacteria communities and PERMANOVA was used to test the significance of explanatory variables.
A Benjamini-Hochberg correction was applied to the Spearman rank correlation analyses.
Results
Physicochemical and biological conditions
The results of the hydrographic surveys showed that downwelling conditions prevailed during winter and spring (Figures 1B; S1–S2). During this period, the important vertical mixing and reduced light availability prevented phytoplankton blooms and relatively high concentrations of inorganic nutrients were measured in surface waters (NO3-, NO2-and SiO2 concentrations and the N:P ratio during winter and spring at both stations were significantly higher than those measured in summer, Mann-Whitney U Test, p-value < 0.05; Table S2; Figure S3). At the coastal station, a persistent surface halocline associated with rain and terrestrial runoff was observed during winter and spring (during winter and spring surface salinity was significantly lower than during summer, Mann-Whitney U Test, p-value < 0.05) (Figures S1, S3; Table S2). By contrast, during summer, the upwelling brought deep, salty, and nutrient-rich water to the surface. In summer, a massive phytoplankton bloom was observed at the coast and spread to the offshore station: at the surface significantly higher Chl-a concentrations accompanied by significantly higher bacterial abundance were measured compared to spring and winter (Mann-Whitney U Test, p-value < 0.05) (see satellite images, Figure S4). The observed very low nutrients concentrations during summer in surface waters at the coastal station are probably due to the massive consumption of nutrients by the phytoplankton bloom (Figures 1B; S1–S3; Table S2). At the coastal station, pronounced changes in Chl-a concentration were observed at the end of the winter cruise (Figures 1B; S1, S5) and even more so, at the end of the summer cruise (likely due to the rapid wind relaxation that interrupted the upwelling and disrupted the bloom (Figure S1), see Joglar et al., 2020 (Joglar et al., 2020). Collectively, there was more daily variation in physicochemical and biological oceanographic conditions (e.g. salinity, nutrients, chlorophyll a and bacterial biomass) in the coastal compared to the offshore and in summer compared to winter (Figures S1–S3, S5; Table S2). An interesting exception to this was temperature, which registered a similar large daily variation in the offshore and coastal stations in summer due to the intense upwelling of cold water that precisely promoted the changes in the rest of variables.
The eukaryotic community at both stations was dominated by dinoflagellates and diatoms (Dinophyceae, Thalassiosira, and Chaetoceros, particularly in summer) (Figure S6A) (Joglar et al., 2020). Among the bacteria, Pelagibacterales and Synechococcales showed higher relative proportions at the offshore station, particularly in winter, while other Alphaproteobacteria and Flavobacteria reached higher relative abundances in spring and summer on the coast. At both stations, Oceanospirillales were relatively more abundant in winter, whereas Cellvibrionales increased in summer (Figure S6B) (Joglar et al., 2020).
Relationships between metatranscriptomes and environment variables
Our metatranscriptomics analysis showed a dominance by Alphaproteobacteria (mainly Pelagibacterales and Rhodobacterales), Gammaproteobacteria (including Alteromonadales and Cellvibrionales), and Bacteroidota (mainly Flavobacteriales) in both stations and in the three sampled seasons (Figure 2A). The taxonomic distribution of transcripts for functional genes was similar in winter and spring, except for higher levels of Cyanobacteria at the offshore station in winter. During the summer phytoplankton bloom at the coastal station there was an important increase in the relative transcription levels of Cellvibrionales (Figure 2A). A relatively higher representation in the metatranscriptomics than in the DNA-based 16S rRNA gene amplicon data was observed for both Pelagibacterales (offshore station) and Other Gammaproteobacteria and Cellvibrionales (coastal station) during the three different seasons (t-test, p-value < 0.05) (Figure S7).
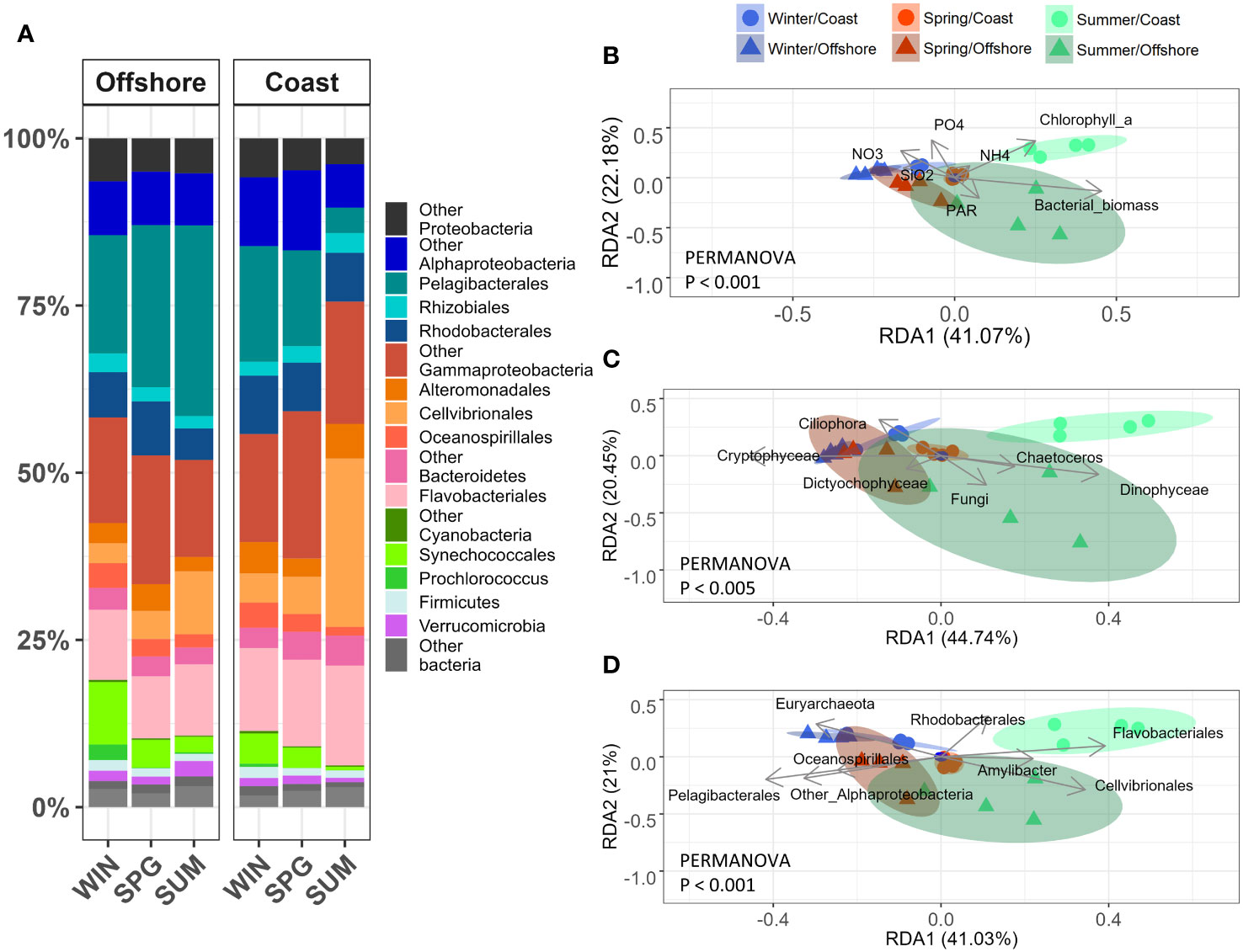
Figure 2 (A) Contribution of the major taxonomic groups to the bacterial metatranscriptome at surface (5m) during winter (WIN), spring (SPG) and summer (SUM) at the coastal and offshore stations. The mean of four replicates (days 1, 3, 5 and 7) per station and season is shown. Redundancy analysis (RDA) of bacterioplankton metatranscriptome data constrained by: (B) environmental variables, (C) relative abundances of main eukaryotic groups (18S), and (D) prokaryotic groups (16S). The metatranscriptome data was clr-transformed to calculate Euclidean distances and to perform the RDA. A PERMANOVA analysis (p-values provided in each plot) was conducted to determine the significance of the analysis. Black arrows indicate the variables that significantly constrain each model. The percentages representing variability are displayed on the axes. Circles: coast, triangles: offshore. Winter: blue, spring: red and, summer: green.
Analysis of Non-parametric MultiDimensional Scaling (nMDS) and PERmutational Multivariate ANalysis Of VAriance (PERMANOVA) of bacterioplankton metatranscriptomes revealed a grouping of samples based on seasons and sampling stations (PERMANOVA, p-value < 0.05) (Figure S8). Redundancy Analysis (RDA) showed that functional profiles of winter and spring bacterial communities from both stations were significantly (PERMANOVA, p-value < 0.001) associated with high concentrations of specific nutrients (NO3- and SiO2) and with fairly high relative abundances (estimated from DNA-based 16S rRNA gene amplicon data presented in Figure S6) of the eukaryotic phytoplankton Ciliophora, Cryptophyceae, and Dictyophiceae (PERMANOVA, p-value < 0.005) and the bacteria Oceanospirillales, Pelagibacterales, and Alphaproteobacteria (PERMANOVA, p-value < 0.001) (Figures 2B–D). During the summer bloom, the bacterial functional profiles showed higher variability and appeared to be linked to increased bacterial abundance, Chl-a concentration, PO3, and NH4+ concentration (PERMANOVA, p-value < 0.001) as well as with Chaetoceros, Dinophyceae, and Fungi at both stations (PERMANOVA, p-value < 0.005) (Figures 2B–D). At the offshore station, bacterial functioning during summer was associated with high relative abundances of Amylibacter, Flavobacteriales, and Rhodobacterales (PERMANOVA, p-value < 0.001), while at the coastal station, it was associated with higher numbers of Cellvibrionales (PERMANOVA, p<0.001) (Figures 2B–D).
Spatial-temporal variability in expression of ecological marker genes
We next assessed the microbial functions related to nutrient fluxes (C, N, P, S, and Fe) and to vitamin synthesis, response to stress and stimuli, and phototrophy in this upwelling system. Different seasonal (Figures 3; S9–S17), daily (Figures 4; S18–S23) and spatial patterns were observed in the different functions studied.
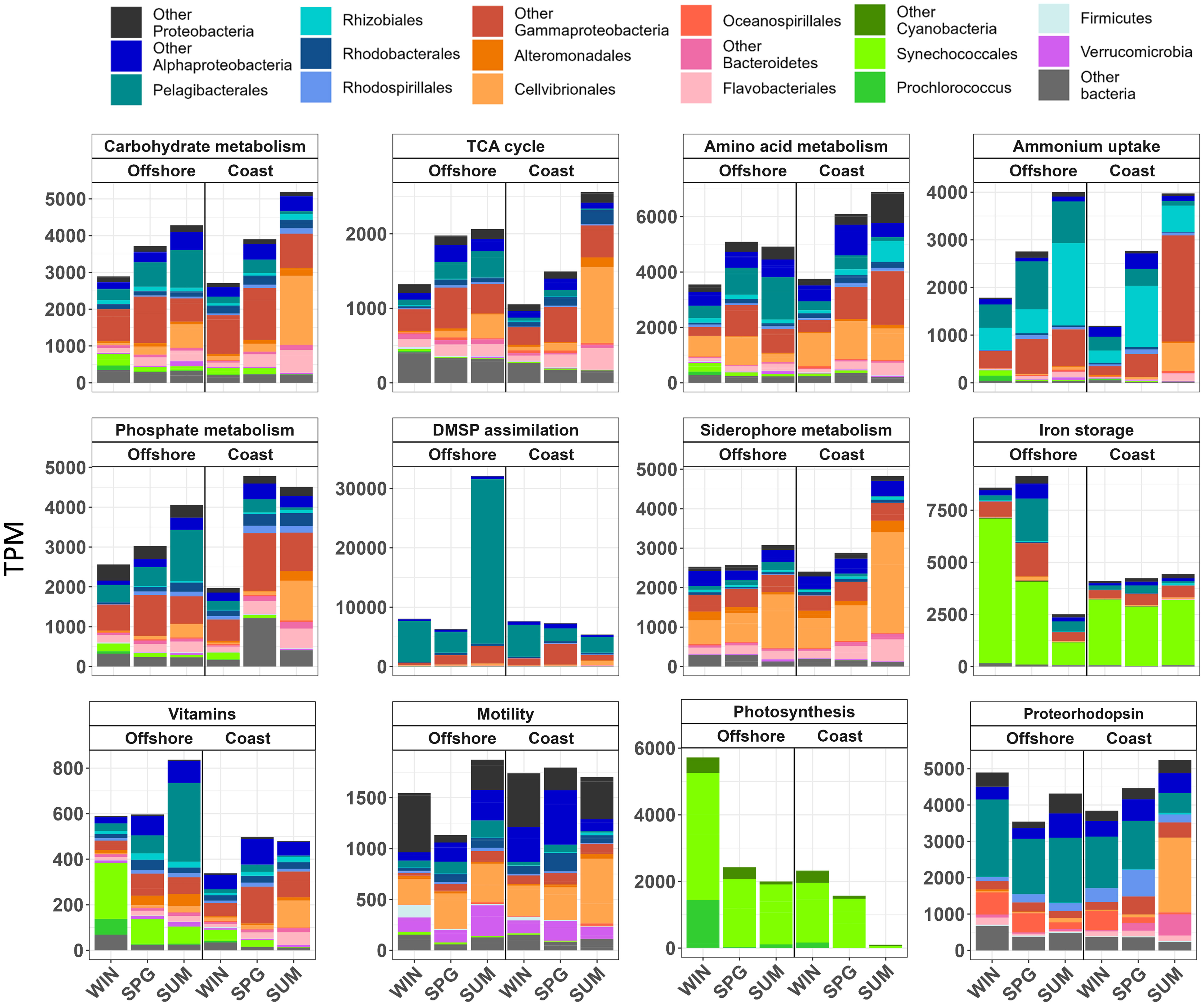
Figure 3 Relative transcript abundance (TPM) of selected key biological process: carbohydrate metabolism, TCA cycle, amino acid metabolism, ammonium uptake, phosphate metabolism, DMSP assimilation, siderophore metabolism, iron storage, vitamins, motility, photosynthesis, and proteorhodopsin. Color bars represent the relative contribution of main taxonomic groups to transcription at the costal and offshore station during winter (WIN), spring (SPG), and summer (SUM). The mean of four replicates (days 1, 3, 5 and 7) is shown. Note that different number of genes are included in each category (see supplementary material) and that different scales are used on the y-axis.
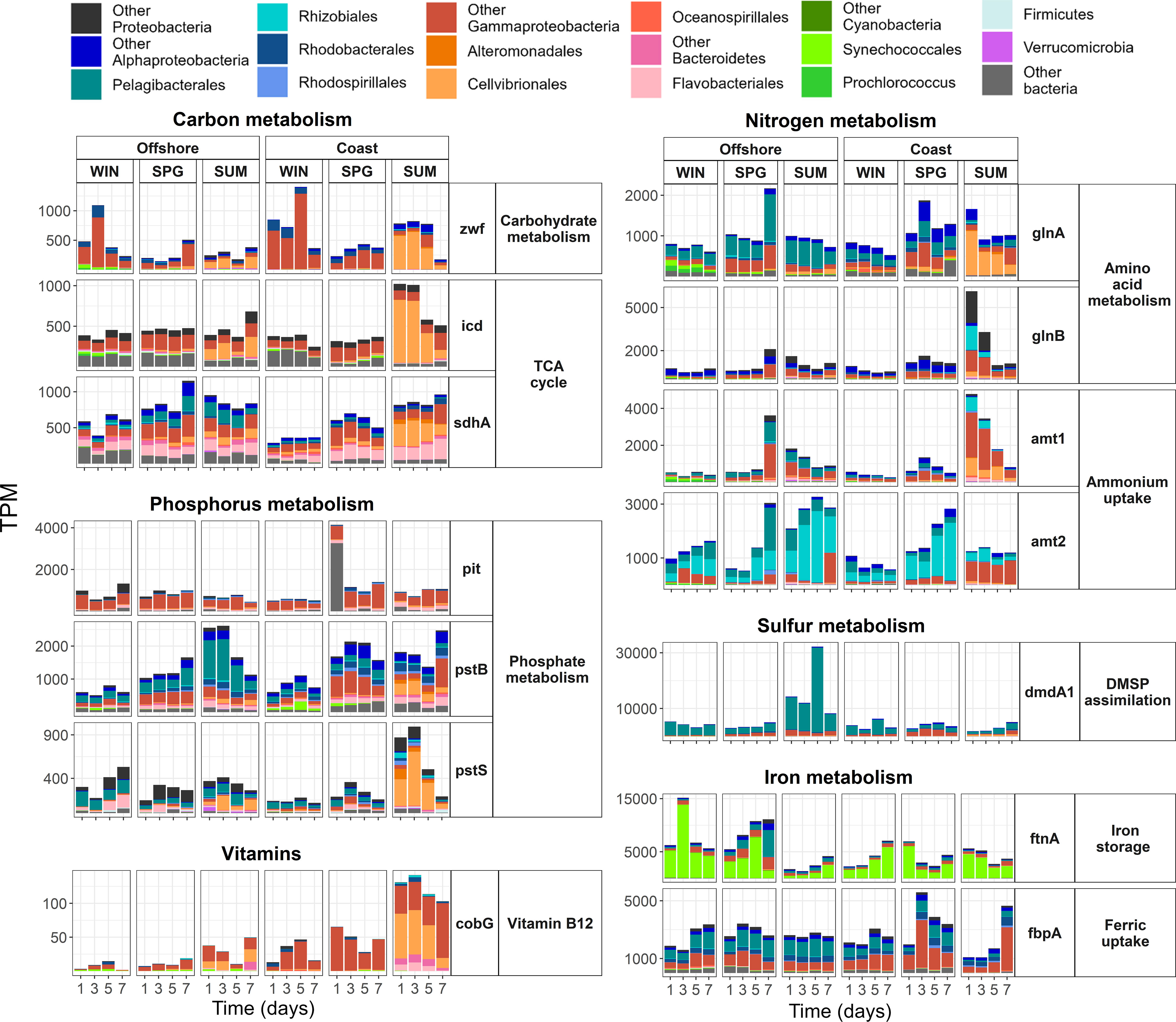
Figure 4 Daily variability of selected genes at the costal and offshore stations during winter (WIN), spring (SPG), and summer (SUM). Relative transcript abundance (TPM) of selected key biological process is provided. Color bars represent the relative contribution of main taxonomic groups to transcription. The value for each sampling day (days 1, 3, 5 and 7) is shown. Note that different scales are used on the y-axis.
Carbon, nitrogen and phosphorous metabolisms
In the carbon metabolism category, high relative transcription of carbohydrate metabolism and TCA cycle genes, particularly by Cellvibrionales and some Alphaproteobacteria, was observed in both stations as the seasons progressed, with higher values in summer during the phytoplankton bloom (Figures 3; S9, S18). Isocitrate dehydrogenase (icd) and succinate dehydrogenase (sdhA) were the TCA cycle genes with highest relative transcription (Figures S9, S18). While icd, (which produces 2-oxoglutarate, also a precursor in amino acid metabolism, see below) was mainly transcribed by Gammaproteobacteria (Cellvibrionales), sdhA was transcribed by several taxa, including Bacteroidetes (Flavobacteriales) and Gammaproteobacteria at the coast and Alphaproteobacteria (Pelagibacterales) at the offshore (Figures S9, S18).
In the nitrogen metabolism category, ammonium uptake and amino acid metabolism were the subcategories with the highest proportion of transcripts, while urea utilization and nitrogen fixation remained low (Figures 3; S10, S18). There were pronounced seasonal changes in the relative expression of ammonium transporter (amt) genes (increasing from winter to summer, when low ammonium concentrations were measured during the phytoplankton bloom in the coast) and different taxa contributed to the transcription of amt-1 (mainly Cellvibrionales) and amt-2 (Pelagibacterales and Rhizobiales) (Figures S10, S18). The dominant genes in amino acid metabolism were also involved in nitrogen assimilation, such as the glnA gene, also importantly transcribed by Cellvibrionales and encoding glutamine synthetase that is the key enzyme that catalyzes the incorporation of ammonium or amino groups from amino acids into glutamine, and the global nitrogen-metabolism regulatory protein P-II (glnB) that modulates the expression of both glutamine synthetase and ammonium transporter genes (Figures S10, S18).
The phosphorus metabolism category was dominated by the expression of genes for phosphate metabolism, with higher relative values in spring and summer than in winter, especially at the coast (Figures 3; S11, S18). Pelagibacterales and Rhodobacterales (offshore and coastal station, respectively) and Gammaproteobacteria (particularly Cellvibrionales in summer at the coast) were the main groups accounting for this expression (Figures 3; S11, S18). Thus, phosphate import ATP-binding protein (pstB) was mainly transcribed by Pelagibacterales at the offshore station and by Rhodobacterales and Cellvibrionales at the coast and the transcription (mainly by Cellvibrionales) of the high affinity phosphate transporter pstS relatively increased in the coast in summer, while the low-affinity phosphate transporter (pit), which peaked in spring at the coast, was mainly transcribed by Gammaproteobacteria at both stations (Figures S11, S18).
Several of the C, N and P metabolism genes at both stations showed important daily changes in relative transcription levels over the course of the 7-day cruises (particularly during summer at both stations, Figures S21–S23) coinciding with short-term oceanographic variability (Figures 5; S5). As an example, individual phosphate acquisition genes showed different dynamics during the last part of the summer cruise at the coastal station (i.e., pstB increased while pstS decreased) and during winter and spring at the coastal station (i.e., pit, pstB, pstS) (Figures 4; S21), coinciding with changes in phosphate availability in the field (Figures 1; S5). Interestingly, daily changes (calculated as the ratio of the daily value divided by the mean value during the four days of the specific season and station) in PO4-3 concentration observed in the field were inversely correlated with the day-to-day changes in dmdA1, amt1, amt2, glnA, pstS and pstB (p < 0.05) (Figure 5). A similar (and counterintuitive) pattern was observed between the daily changes in these genes and those in the N/P ratio (Figure 5). It is important to note here that daily changes in the N/P ratio were mostly driven by changes in NO3- concentrations and not in PO4-3 availability (Figure 1B). On the other hand, daily changes in bacterial biomass were inversely correlated with the day-to-day changes in icd, zwf and amt2 and directly correlated with those in sdhA, pit, dmdA1 and fbpA (p < 0.05) (Figure 5). Finally, daily changes in temperature were directly correlated with the day-to-day changes in icd, zwf, amt1, amt2, glnA, glnB and pstS and inversely correlated with those in pit (p < 0.05) (Figure 5).
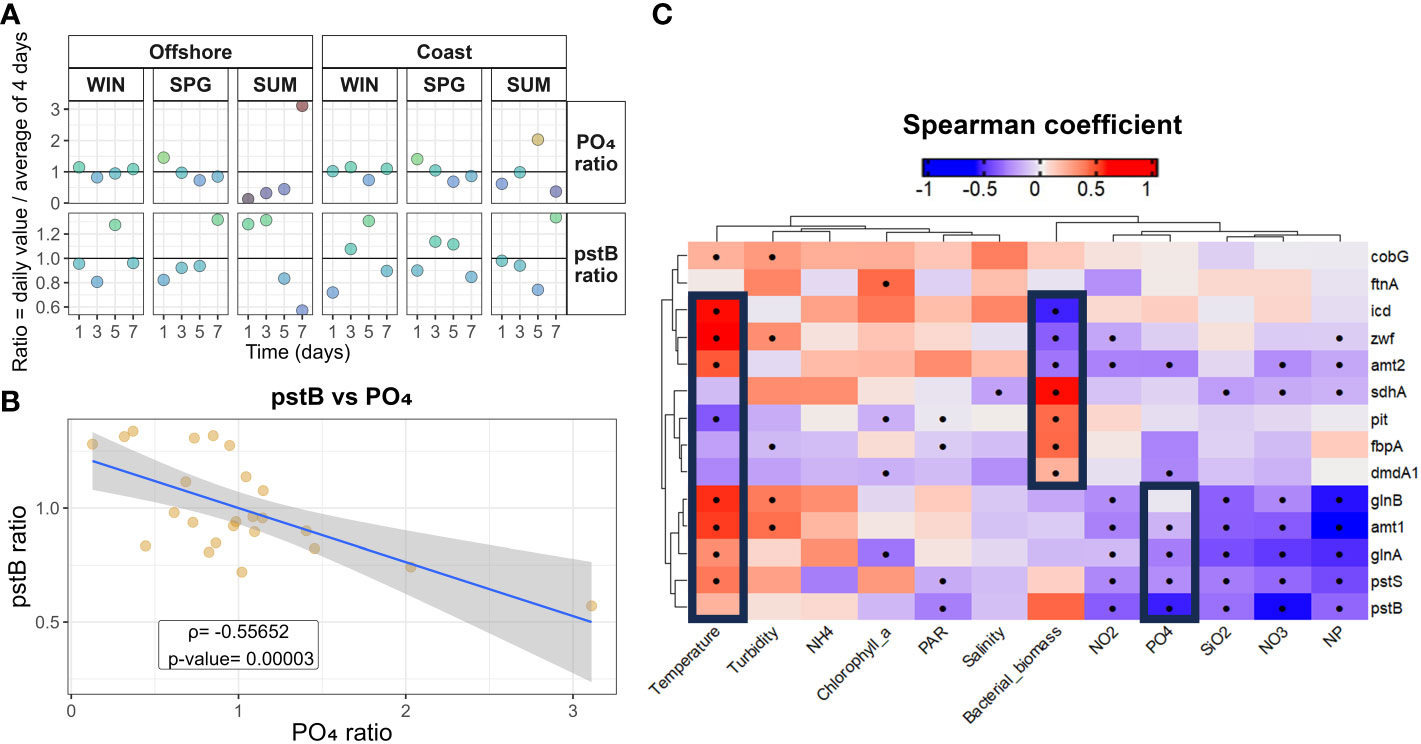
Figure 5 (A) Daily variability (estimated as the ratio of the daily value of each variable divided by the mean value of that variable during the four days of the specific season and station) of phosphate (PO₄³⁻) and of pstB transcripts measured at the costal and offshore stations during winter (WIN), spring (SPG), and summer (SUM). The color and the vertical position of the bubble (y-axis) represent the value of the ratio (B) Correlation between values presented in panel (A), Spearman coefficient and p-value of the correlation are provided. (C) Spearman rank correlation analysis between daily variability of the different environmental variables and that of selected genes. A Benjamini-Hochberg method was used to adjust the p-values. Dots indicate p < 0.05. NP stands for N:P ratio.
Sulfur, iron and vitamin metabolisms
Dimethylsulfoniopropionate (DMSP) assimilation, especially Pelagibacterales dmdA1 and dmdA2 transcription, was the category with the highest relative expression levels of all categories studied (Figures 3, 4; S12). A large peak in DMSP assimilation was observed in summer at the offshore station coinciding with a relative increase in the contribution of Dinophyceae to the eukaryotic community (Figure S6). Large short-term changes were observed in the transcription of DMSP assimilation genes during the final phase of the summer cruise at both the coastal and offshore stations (Figures 4; S19, S22). Relative expression of genes for sulfite and sulfate assimilation also reached fairly high levels, accounted for by Pelagibacterales and Gammaproteobacteria for sulfite (e.g., adenylsulfate reductase genes aprAB) and by a variety of taxa for sulfate (e.g., cysN) (Figure S12). Synechococcales and Cellvibrionales increased their relative contribution to cysN transcription in winter at the offshore station and during the summer bloom at the coastal station, respectively (Figure S12).
Iron metabolism genes were transcribed by various taxa, including Cellvibrionales, Flavobacteriales, and Synechococcales (Figures 3, 4; S13). The relative transcription of iron storage genes increased at the offshore station during winter and spring. In contrast, pronounced relative increases in siderophore transport (e.g., exbB by Cellvibrionales), and ferric uptake were observed at the coast during summer and spring, respectively (Figures 3, 4; S13). The expression of ferric ion uptake genes (e.g., fbpA) was mainly associated with Pelagibacterales, while iron storage genes (e.g., ftnA) were mostly associated with Synechococcales (Figures 3, 4; S13). Interestingly, day-to-day changes in expression of iron uptake (i.e., fbpAB) and storage (ftnA) genes during the summer bloom at the coastal station showed opposite patterns (Figures 4; S19, S22).
Vitamin synthesis marker genes selected were on one hand, thiL and thiC as marker genes of B1 synthesis metabolism and, on the other hand, cbiX (as marker gene of the anaerobic pathway) and, cobG, cobN and cobT (as marker genes of the aerobic pathway) for B12 synthesis metabolism. Vitamin synthesis genes generally showed higher relative expression levels in summer at the offshore station and during spring and summer at the coastal station (Figure 3). Vitamin synthesis genes were mainly related to Alphaproteobacteria (mostly Pelagibacterales at the offshore station), Gammaproteobacteria (particularly Cellvibrionales at the coastal station) and Synechococcales and Flavobacteriales at the offshore and at the coastal station, respectively (Figure 3). Specific genes related to vitamin synthesis presented different spatial-temporal patterns and were transcribed by different taxonomic groups (Figures 4; S14). Still, there were marked spatial-temporal patterns for different vitamins and bacterial taxa. For example, vitamin B1 gene thiL and B12 gene cobG were mainly transcribed by Cellvibrionales and Flavobacteria, with increased relative transcription in summer, particularly at the coastal station (Figures 4; S14, S19). Pelagibacterales instead dominated the transcription of vitamin B12 gene cobT, particularly at the offshore station during summer (Figures S14, S19). By contrast, vitamin B12 gene cbiX was transcribed by multiple taxa with no clear patterns (Figures S14, S19). Highest daily variability in the relative transcription of genes related to vitamins was observed during winter at the offshore station (Figure S22).
Motility, phototrophy and response to stress
Transcription of stress response genes was distributed among Pelagibacterales (offshore) and Cellvibrionales (coast) and increased at the coastal station in summer and at the offshore station in spring (Figures S15, S20). The expression of stimuli-related genes (e.g., motility genes like fliC) was mostly associated with Cellvibrionales, Alphaproteobacteria, and Verrucomicrobia. (Figures S16, S20). Highest daily variability in the relative transcription of genes related to response to stress and stimuli were observed during summer at both stations (Figure S22).
In the phototrophy category, the relative transcription of our selected photosynthesis genes (up to ~5500 TPM; Figure 3) was dominated by Synechococcales and was generally higher at the offshore station where it peaked in winter (Figures 3; S17, S20). This included genes for light harvesting antenna (e.g., cpcAB for phycocyanin), the photosystems (psa and psb) and carbon fixation (rubisco subunits). In contrast to the variability in photosynthesis genes, the proteorhodopsin (prd) gene expression was relatively stable and high (~4000) across all samples (Figure 3). The prd expression was generally dominated by Pelagibacterales with fair contributions also by other taxa - especially Cellvibrionales at the coastal station in summer (Figures 3; S17, S20). Bacteriochlorophyll gene (puf) expression was below 5 TPM (Figures S17, S20). Highest daily variability in the relative transcription of genes related to phototrophy was observed during winter at the offshore station (Figure S23).
Relationships between bacterial functioning and phytoplankton community composition
Since eukaryotic phytoplankton are key components of upwelling ecosystems – potentially shaping the conditions for bacterial activities – we carried out Spearman rank correlation analyses (applying a Benjamini-Hochberg correction) between the relative abundance of the major eukaryotic taxa in the 18S rRNA gene amplicon data and the relative transcription levels of the studied prokaryotic functional categories (Figure 6A) and environmental variables (Figure 6B). Notably, bacterial biomass, temperature, and salinity displayed significantly positive correlations (p-value < 0.05 marked with asterisks in Figure 6) with categories related to bacterial uptake and utilization of resources (e.g., metabolism of carbohydrates, amino acids, phosphate and sulfate, and ammonium uptake) while the opposite pattern was observed for nutrients concentration (p-value < 0.05 Figure 6B). Significant positive correlations were also found between the categories related to bacterial uptake and utilization of resources and the relative abundances of Stramenopiles (particularly Chaetoceros), Dinophyceae, marine stramenopiles (MAST), and Fungi (p-value < 0.05) (Figure 6A). A positive relationship was found between the transcription of DMSP assimilation genes by the prokaryotic community and the abundance of Dinophyceae (p-value = 0.052) and marine alveolate (MALV) (p-value < 0.05) (Figure 6A). In contrast, other metabolic traits, such as sulfite assimilation, iron storage, and photosynthesis genes, showed a significantly positive correlation (p-value < 0.05) with Chlorophyta (particularly Ostreococcus and Micromonas) and Cryptophyceae (Figure 6A) and with low chlorophyll a values (p-value < 0.05) (Figure 6B).
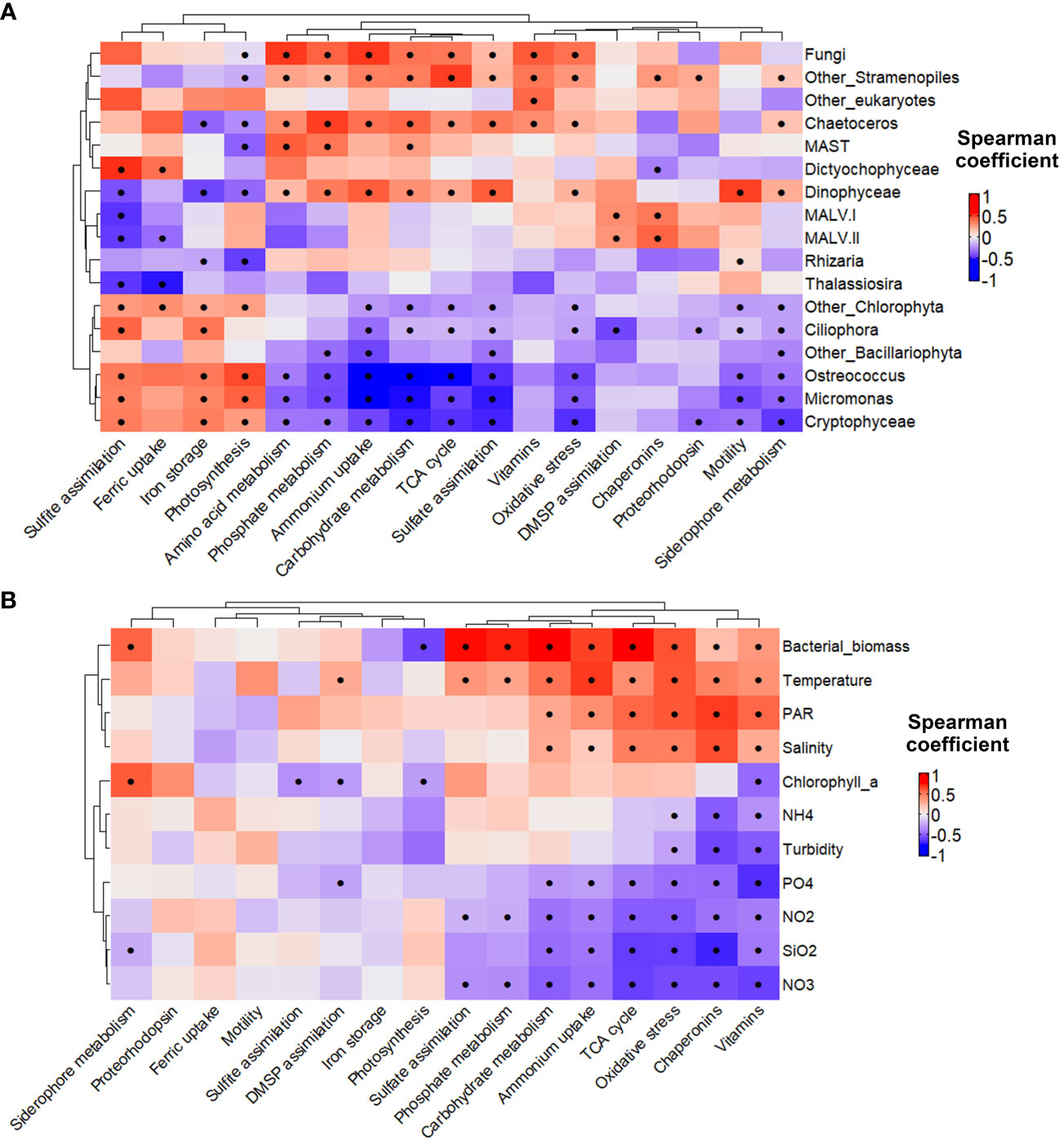
Figure 6 Spearman rank correlation analysis between transcription of selected prokaryotic gene subcategories and (A) the relative abundance of the major eukaryotic taxa (18S) and; (B) environmental variables. The metatranscriptome data was clr-transformed before the analysis and a Benjamini-Hochberg method was used to adjust the p-values. Dots indicate p < 0.05.
Discussion
Coastal upwelling systems: dynamic grids of bacterioplankton functional specialization
Our analyses provided novel information about the potential mechanisms governing microbial interactions, nutrient utilization, and energy and matter fluxes in a representative upwelling system. We reason that it is essential to identify and characterize the large diversity of microbial processes and patterns for two important reasons. First, it partly results from two-way interactions with the inherently diverse distribution patterns of phytoplankton and bacteria within the microbial communities. Second, because of the complex interdependency between different physicochemical conditions and the myriad of ecological processes driven by different microbes in the ocean. Thus, focusing on the transcriptional investment by any one taxon in any particular metabolic pathway or a specific season of choice, would unavoidably have resulted in underestimating the magnitude of dynamics and distribution patterns characteristic of coastal upwelling systems. In the following we will discuss these findings with the intention to understand potential linkages between physicochemical and biotic processes.
Resource availability as a powerful driver in structuring microbial functioning
The present work is, to our knowledge, the first studying high resolution spatial and temporal changes in bacterial functioning by means of metatranscriptomics in an upwelling system. The coastal area of the NW Iberian Peninsula is a highly dynamic ecosystem, affected by the intermittent upwelling of cold and inorganic nutrient-rich water that promotes phytoplankton and bacterial growth (Figueiras and Ríos, 1993; Nogueira et al., 1997; Álvarez-Salgado et al., 2002). Our dataset suggests that the coast to offshore gradient and the temporal changes in environmental and biological conditions promoted by the upwelling are key drivers of the observed pronounced taxonomic and functional bacterial diversification in this system. Several examples extracted from our results may be given.
The present dataset exemplifies some specific functional mechanisms behind the important role of nitrogen (N) and phosphorus (P) in structuring the functioning of microbial communities previously suggested to exist in this ecosystem (Martínez-García et al., 2010; Teira et al., 2016). Regarding nitrogen, the observed high expression of the amt, glnAB, and icd genes by Gammaproteobacteria (particularly Cellvibrionales) during summer at the coastal station was remarkable. The enzymes encoded by these genes are linked in the process of nitrogen assimilation from ammonium in that amt encodes an ammonium transporter, glnAB encode glutamine synthase that synthesizes glutamine from ammonium and glutamate (glutamine can next be used for amino acid synthesis), and icd encodes the TCA cycle enzyme isocitrate dihydrogenase that produces 2-oxoglutarate, which is the precursor of glutamate. This may suggest that the competitive ability of Cellvibrionales during the phytoplankton bloom in part lies in their ability to gear their metabolism towards utilizing N from ammonium as a means to utilize the C that they process. This is in line with the emerging view that the dialogue between the metabolic N and C cycles is largely mediated at the intercept between the TCA-cycle and the synthesis of amino acids (Forchhammer et al., 2022). At the offshore station, on the other hand, Pelagibacterales showed a relatively higher transcription of ammonium transporters compared to other taxa, suggesting this group is more capable of assimilating ammonium at the low concentrations typically found in oceanic areas. We think analysis of adjustments of central metabolism between biosynthetic as compared to energy-yielding pathways (such as respiration) has a large potential to inform on how marine bacteria meet challenges in elemental stoichiometry of their resources (as imposed through e.g. C- or N-limitation). Especially if advances are made in characterizing gene systems like amino acid transporters and extracellular proteases involved in the utilization of dissolved organic nitrogen.
The high expression of the gene that encodes the transport system for orthophosphate (pstB) during summer, when phosphate concentration was low, is coherent with the suggested role of the Pst system (pstABCS) in inorganic phosphate (Pi) uptake only under Pi-limiting conditions (Harke and Gobler, 2013; Jimenez-Infante et al., 2017). On the other hand, the transcription of the pit gene, a low-affinity phosphate membrane transporter typically expressed under high phosphorus concentrations and post-bloom conditions (Alonso-Sáez et al., 2020), peaked at the coast in spring, coinciding with the highest ammonium and phosphate concentrations registered. Note here that, even though the phosphate concentration was similar in winter and spring at the coastal site, dissolved inorganic N, particularly ammonium, was considerably higher in spring, suggesting that higher expression of the pit gene in spring than in winter might be related to the higher ammonium to phosphate ratio. Overall, the different spatial-temporal patterns of variability observed in genes with different phosphate affinity in this upwelling system, regardless of the relatively limited variability in phosphate ambient concentration, reinforces the hypothesis of a complex role of phosphate in structuring bacterial functioning (Harke and Gobler, 2013; Satinsky et al., 2014).
Our results support the idea of a taxonomic diversification in iron (Fe) provision strategies in this upwelling system. Thus, while Cellvibrionales and Flavobacteriales are suggested to mostly use siderophore-associated Fe, Pelagibacterales probably use ferric ions and Synechococcales accumulate Fe as ferritin in this ecosystem (Ahlgren et al., 2020). On the other hand, a widespread genetic capacity for the uptake and utilization of sulfate in surface waters off NW Iberian Peninsula can be deduced from our data, as opposed to previous knowledge limiting sulfate use during phytoplankton blooms to a few bacterial groups (Zhou et al., 2020). Interestingly, our results also highlight a probable dominant role of Pelagibacterales in sulfite assimilation in the system, extending to upwelling areas previous findings in offshore epipelagic layers (Needham et al., 2017). The present dataset suggests for the first time the relevant role of organic sulfur (S) compounds as sources of reduced S for marine prokaryotes in this upwelling system, confirming previous studies in other areas (Kiene et al., 2000; Moran et al., 2003). Interestingly, the high relative abundance of dmdA transcripts at the offshore station during the summer upwelling period coincided with a dominance of the phytoplankton community by Dinophyceae. Dinoflagellates are known to have high dimethylsulfoniopropionate (DMSP) production rates (Zhao et al., 2021), and previous work has suggested that their DMSP-producing ability could affect the structure of their associated bacterial community (Lin et al., 2021). In this regard, the observed dominant role of Pelagibacterales in the transcription of DMSP utilization genes expands the work by Varaljay et al. (2012) in the North Pacific subtropical gyre to this upwelling system. Furthermore, DMSP has been previously reported to have a photoprotective function (Archer et al., 2009), which may be related to the high radiation levels and the strong thermal stratification registered in the offshore station during summer.
Finally, the present dataset suggests that different strategies for using light as a source of energy by bacteria occur in this upwelling system. Thus, the relative transcription of photosynthesis related genes by Synechococcales was more important in the offshore station during winter, where the abundance of this group was relatively higher (Joglar et al., 2020). It is important to note here that, as explained in the Methods section, the coastal and offshore stations were sampled at different time points in the daily cycle. Thus, the high relative transcription of photosynthesis-related genes by Synechococcales during winter in the offshore station may be not only related to the broad differences in physicochemical and biological conditions (higher relative abundance of Synechococcales during winter in the offshore station, see Figure S6) but also partly to an increase in the investment in light capture due to the sampling time. In this regard, previous studies have shown that prokaryotic transcriptomes from natural samples may exhibit diel periodicity, particularly those of Cyanobacteria in open ocean waters (Ottesen et al., 2013; Ottesen et al., 2014). Interestingly, the data on the transcription of the prd gene suggest that photrophy mediated though proteorhodopsin was relevant and widespread among different taxa, including Pelagibacterales, in accordance with the results by Olson et al. (2018) in oligotrophic waters and Gammaproteobacteria during the coastal phytoplankton bloom.
Short-term dynamics in microbial functioning
Previous studies have shown recurring daily patterns in the functioning of heterotrophic bacteria from relatively stable oligotrophic environments (Ottesen et al., 2014; Muratore et al., 2022). It is also well known that the metabolism of naturally occurring bacteria experience inter- and intra-seasonal changes associated to phytoplankton blooms (Teeling et al., 2012; Teeling et al., 2016). However, this is to our knowledge the first time that systematic changes in specific microbial functions (identified from gene expression analyses) associated to short-term oceanographic variability are shown. Our results evidence important short-term dynamics in bacterioplankton functioning superimposed on seasonal variability that coincided with changes in hydrography, nutrient availability and biotic variables in this system. Interestingly, this day-to-day changes in transcripts for specific functions related to nutrient fluxes and response to stimuli were even more pronounced when daily variability in oceanographic conditions (e.g. temperature, phytoplankton biomass, resource availability) was highest, that is: close to the coast during summer.
In summer, a wind relaxation event interrupted the ongoing upwelling, which modified the vertical density gradient at the coastal station, altered the hydrographic conditions and nutrient availability, and disrupted the phytoplankton bloom (see (Barbosa et al., 2001) and (Joglar et al., 2021b)). Concomitantly, a sharp decrease in the transcription of functions related with bacterial utilization of resources derived from the phytoplankton bloom was observed, including amino acid metabolism (glnAB), TCA cycle (icd), and ammonium uptake (amt). This suggests that bacteria quickly responded to the changes in resource supply and potentially also in the C/N ratio of resources, as previously suggested by others (see review by Forchhammer et al. (2022)). Also, the relative importance of high-affinity phosphate uptake genes (e.g., pstS and pstB, which are considered an indicator of microbial P limitation and that varies seasonally in relation to P availability (Dyhrman et al., 2007)) decreased quickly after bloom disruption (on day 5), this probably due to the cease of the competition with phytoplankton. In contrast, the transcription of low-affinity phosphate uptake gene pit followed the opposite pattern and relatively increased at the end of the summer sampling at the coast when phosphorus availability increased. Similarly, bacteria shifted from a strategy based on iron storage to an increase in the relative importance of iron uptake (as suggested by the high relative transcription of ftnA and fbpAB, respectively) when the phytoplankton bloom was interrupted. Interestingly, the important short-term changes observed in the transcription of DMSP assimilation genes coincided with day-to-day peaks in the relative abundance of dinoflagellates during the final phase of the summer cruise at both stations. Overall, these results suggested a close coupling between the physicochemical conditions and the bacterial and algal component of microbial communities from this upwelling system.
Unraveling functional interactions between bacteria and coastal phytoplankton
Overall, our results demonstrate that specific functional processes of the prokaryotic community identified from gene expression analyses) are associated with different phytoplankton groups in the upwelling system off the NW Iberian Peninsula. The presence of different phytoplankton and bacterioplankton populations during periods with different environmental conditions has been suggested in previous studies both in the area (Joglar et al., 2020; Costas-Selas et al., 2023) and in other marine ecosystems (Pinhassi et al., 2004; Needham et al., 2017; Bunse et al., 2019). Recent work in the NW Iberian Peninsula suggests a key role of biotic interactions as structuring factors of the eukaryotic community, mostly driven by positive associations between phytoplankton and bacteria (Costas-Selas et al., 2023). These findings may suggest some coevolution between the algae and the associated bacterial community that extends beyond the ability to transform phytoplankton-derived macromolecules by bacteria. The present dataset unravel the specific metabolic processes and bacterial and phytoplankton taxa that may be behind these associations through space and time. It is important to note here again that the interactions between phytoplankton and bacteria may change throughout the light cycle as it is known to affect the functioning of photosynthetic organisms (Ottesen et al., 2013; Ottesen et al., 2014). Therefore, the sampling at different times in both stations might influence to some extent the results discussed below.
Two interesting examples of possible phytoplankton-bacteria linkages observed during the productive season were associated to the bacterial transcription of genes related to the metabolism of DMSP (see above) and to the synthesis of B-vitamins. The observed increase in the relative abundance of transcripts of vitamin-related genes during summer is likely related to the high phytoplankton and bacterial requirements characteristic of blooming populations (Croft et al., 2005; Koch, 2012; Sañudo-Wilhelmy et al., 2012; Koch and Trimborn, 2019). Interestingly, genes belonging to the aerobic pathway of B12 synthesis (e.g. cobG and cobT), which is mostly associated with phytoplankton blooms (Zhou et al., 2020), increased during summer, while transcription of cbiX, which belongs to the anaerobic pathway of B12 synthesis that has been previously suggested to be independent of phytoplankton activity (Grossman, 2016), did not follow a clear seasonal pattern. The relative increase in thiL and cobG transcription by Cellvibrionales and Flavobacteria during summer suggests that these groups provided B1 and B12 vitamins to blooming eukaryotic phytoplankton. Interestingly, this hypothesis is also supported by microcosms experiments stimulating phytoplankton growth and including B1 and B12 vitamins additions carried out in summer in the NW Iberian Peninsula upwelling system that showed relatively important contributions of Cellvibrionales and Flavobacteria to the transcription of genes related to vitamin metabolism (Joglar et al., 2021a).
Finally, our results suggest strong functional linkages between bacteria and phytoplankton during non-blooming winter conditions. In this regard, the association between transcripts related to bacterial metabolic traits such as sulfite assimilation, iron uptake and storage, and photosynthesis, and Clorophyta and Cryptophyceae may reflect the close association between these phytoplankton groups and bacteria from the orders Pelagibacterales and Synechococcales, which played prominent roles in the relative transcription of genes associated with these metabolic functions. The close connection between these phytoplankton groups and Synechococcus sp., also described by Costas-Selas et al. (2023) in the same study area, were hypothesized to be linked with B12 provision. Our results point to an additional interaction mechanism between these groups, which could be related to the high capacity of Synechococcus to store Fe. As Clorophyta and Cryptophyceae may have mixotrophic behavior (Stoecker et al., 2017), predation on Synechococcus might be a source of Fe for them. A previous experimental study observed a sharp decrease in genes related to Fe uptake in a mixotrophic Haptophyta fed with bacteria, suggesting that bacterivory may provide Fe to the algae (Liu et al., 2015).
The close correlation between Chlorophyta (particularly Ostreococcus and Micromonas), Cryptophyceae, and Pelagibacterales suggests a link mediated by the sulfur metabolites, given the strong correlation between these phytoplankton groups and the relative abundance of sulfite assimilation genes. In this regard, a recent study suggests that adenylylsulfate reductase genes (aprAB) in Pelagibacterales clades could be related to taurine metabolism, oxidizing the sulfite derived from the conversion of taurine into acetyl-CoA (De Corte et al., 2021; Ruiz-Perez et al., 2021), a result consistent with the demonstrated ability of Ostreococcus and Micromonas to produce high quantities of taurine (Durham et al., 2019).
Cellvibronales: an unexpected key player in upwelling systems
Knowledge of the ecophysiology and ecology of Cellvibrionales is still scarce, but they appear to reach highest relative abundances in surface waters and coastal areas (Chénard et al., 2019; Pajares et al., 2020; Reji et al., 2020). Although Cellvibrionales is not the most abundant bacterial group in this system, it appears to play a relevant and unexpected role during the summer phytoplankton bloom formation and development.
First, our field data suggest a prominent role of Cellvibrionales in degradation of phytoplankton-derived organic matter. Thus, during the Dinophyceae and Chaetoceros summer bloom, Flavobacteriales and Cellvibrionales dominated the relative expression of genes related to resource utilization and remineralization. It is well known that the primary role of Flavobacteriales in the degradation of polymeric organic matter (Teeling et al., 2012; Fernández-Gómez et al., 2013) is especially important during diatom blooms in upwelling ecosystems (Klindworth et al., 2014). However, the high transcription by Cellvibrionales of carbohydrate and TCA cycle-related genes as well as ammonium utilization and amino acid synthesis genes during the phytoplankton bloom presented here is remarkable. This behavior may be related to efficient energy production (Wang et al., 2022) and to adjustments to C or N-limitation by this group when resources are available during summer phytoplankton blooms (see above). Thus, the increase in the transcription of C metabolism-related genes by Cellvibrionales is consistent with recent findings in concurrent mesocosms experiments (performed in the same sampling area during the present study) (Pontiller et al., 2022) and in other regions (Poretsky et al., 2010; Liu et al., 2020), suggesting a quick response of Cellvibrionales to phytoplankton-derived organic matter. In surface waters of coastal systems like the NW Iberian Peninsula, the Gullmar Fjord and the German Bight, Cellvibrionales have been shown to importantly invest energy in the expression of different polymer degrading enzyme systems including for example laminarases that allow bacteria to decompose the algal storage glucan laminarin (Teeling et al., 2012; Pontiller et al., 2021; Pontiller et al., 2022) emphasizing their significant role in the turnover of phytoplankton-derived organic matter in these systems. Similarly, the relatively abundant Cellvibrionales transcription of motility-related genes during the summer bloom is in accordance with their description as a copiotroph group, known to quickly respond to the increase of DOM surrounding the microenvironment of phytoplankton cells in productive environments (Smriga et al., 2016). In addition, the high transcription of the proteorhodopsin gene (i.e. prd) by Cellvibrionales during summer may indicate that this group uses proton pumps as a supplemental energy source to uptake recently released complex carbon compounds during phytoplankton blooms (Pinhassi et al., 2016).
Our data suggest that the association between blooming phytoplankton and Cellvibrionales may be more complex than just the ability to transform phytoplankton derived macromolecules. We found higher levels of expression of genes for vitamins B1 and B12 synthesis by these bacteria associated with phytoplankton groups dominant during the blooms (i.e., Chaetoceros and Dinophyceae). This finding suggests that Cellvibrionales have an important role as suppliers of micronutrients needed for phytoplankton growth and bloom development. Similarly, the increase in the expression of siderophore-uptake related genes by Cellvibrionales associated with bloom-forming phytoplankton may indicate that these bacteria compete with Chaetoceros and Dinophyceae algae for organically-bound iron during the bloom season (Amin et al., 2009). Therefore, our data suggest that Cellvibrionalles develop a mutualistic relationship with blooming phytoplankton by producing growth-promoting compounds like vitamin B12 and facilitating phytoplankton functioning. They also show, as explained above, that Cellvibrionalles seem to act as competitors of blooming phytoplankton for iron provision in this productive upwelling system.
In conclusion, the present work highlights the mosaic of microbial processes governing energy and matter transformations in this representative upwelling system. Uncovering the linkages between bacterial activity and spatial-temporal variability in environmental conditions represents a relevant goal for microbial oceanography The systematic patterns that emerge from this work are encouraging in the context of future modeling attempts aiming to predict biogeochemical processes in such highly productive ecosystems.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/ena, PRJEB36188 (16S rRNA gene); PRJEB36099 (18S rRNA gene) and; PRJEB36728 (ERS5513557-ERS5513582) (metatranscriptome sequences).
Author contributions
ED-N: Formal analysis, Writing – original draft. ET: Writing – original draft, Conceptualization, Funding acquisition, Writing – review & editing. BP: Writing – original draft, Writing – review & editing, Formal analysis. DL: Formal analysis, Writing – original draft. VJ: Formal analysis, Writing – original draft. CP-A: Writing – review & editing. EF: Writing – review & editing, Conceptualization, Funding acquisition, Writing – original draft. JP: Conceptualization, Writing – original draft, Writing – review & editing, Formal analysis. SM-G: Conceptualization, Formal analysis, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Spanish Ministry of Science and Innovation through the ENVISION (CTM2014-59031-P) INTERES (CTM2017-83362-R), TRAITS (PID2019-110011RB-C33) and BIOTOX (PID2021-125643OB-C21) projects.
Acknowledgments
The authors would like to acknowledge support from Science for Life Laboratory, the National Genomics Infrastructure, NGI, and Uppmax (compute project SNIC 2017/7-419 and storage project SNIC 2020/16- 76) for assisting in massive parallel sequencing and computational infrastructure. We also thank the crew of B/O Ramón Margalef for their hospitality and professionalism during cruises and all the people involved in the project ENVISION for helping with sampling and analytical work. Erick Delgadillo acknowledges a predoctoral FPI fellowship (PRE2018-085871) from the Spanish Ministry of Economy and Competitiveness.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1259783/full#supplementary-material
Supplementary Table 1 | List of the 162 genes found out of the 198 target protein profiles grouped into nine categories and 44 subcategories.
Supplementary Table 2 | Average and standard deviation of the different oceanographic parameters measured at the offshore and coastal stations during winter, spring and summer.
Supplementary Figure 1 | Vertical distribution over time of temperature (°C; panels 1-6), photosynthetically active radiation (µmol photons m2 s-1; 7-12), Chl a derived from calibrated CTD fluorescence sensor (µg L-1; 13-18), salinity (PSU; 19-24) and turbidity (NTU; 25-30) in winter, spring and summer cruises at the offshore and coastal stations.
Supplementary Figure 2 | Vertical distribution over time of ammonium (NH4+ µmol L¹; panels 1-6), nitrite (NO2- µmol L¹; 7-12), nitrate (NO3- µmol L¹; 13-18), silicate (SiO2 µmol L¹; 19-24) and phosphate (PO43- µmol L¹; 25-30) in winter, spring and summer cruises at the offshore and coastal stations.
Supplementary Figure 3 | Box plots of salinity, temperature, chlorophyll a, bacterial abundance, nitrate (NO3-), ammonium (NH4+), phosphate (PO43-), N:P ratio, and silicate (SiO2) at surface (5m) during winter (WIN), spring (SPG) and summer (SUM). Horizontal line in the box represents the median value. The whiskers above and below the box represent the range of the data (maximum and minimum) within 1.5 times the interquartile range (IQR). Points outside the whiskers are outliers.
Supplementary Figure 4 | Chlorophyll-a composite from Aqua-MODIS Composite satellite images during the cruise periods, showing the chlorophyll-a filaments extending form the coast to the ocean during the upwelling in summer. During summer offshore chlorophyll-a is very low due to nutrient-limitation associated to thermal stratification.
Supplementary Figure 5 | Daily variability (estimated as the ratio of the daily value of each variable divided by the mean value of that variable during the four days of the specific season and station) of the different oceanographic variables measured at the costal and offshore stations during winter (WIN), spring (SPG), and summer (SUM). The color and the vertical position of the bubble (y-axis) represent the value of the ratio.
Supplementary Figure 6 | Temporal evolution of the relative abundances of major taxonomic groups of: (A) prokaryotes (16S) and (B) eukaryotes (18S) at surface (5m) in the coastal and offshore stations during winter, spring and summer. Data for each sampling day (days 1, 3, 5 and 7) is shown.
Supplementary Figure 7 | Relative abundance of major prokaryotic groups (16S) (x-axis) and relative contribution of each group to the metatranscriptome (y-axis) in the offshore and coastal stations during winter, spring and summer. For each taxonomic group, the value in the y and x-axis were significantly different (t-test, p<0.05) in all cases except for: Oceanospirillales, Pelagibacterales, Rhodobacterales and Other Alphaproteobacteria in the offshore station in winter; Cellvibrionales, Rhizobiales and Other Alphaproteobacteria in the offshore station in spring; Cellvibrionales and Verrucomicrobia in the offshore station in summer; Oceanospirillales, Pelagibacterales, Prochlorococcus and Other Alphaproteobacteria in the coastal station in winter; Cellvibrionales, Oceanospirillales, Synechoccales and Verrucomicrobia in the coastal station in spring and; Oceanospirillales, Synechoccales, Prochlorococcus and Verrucomicrobia in the coastal station in summer (t-test, p>0.05).
Supplementary Figure 8 | Analysis of Non-parametric MultiDimensional Scaling (nMDS) of clr-transformed bacterioplankton metatranscriptomes. PERmutational Multivariate ANalysis Of VAriance (PERMANOVA, p < 0.01) was used to differentiate between groups. Circles: coast, triangles: offshore. Winter: blue, spring: red and, summer: green.
Supplementary Figure 9 | Relative transcript abundance (TPM) of carbon metabolism-related genes. Color bars represent the relative contribution of main taxonomic groups to the transcription of each gene at the costal and offshore station during winter (WIN), spring (SPG), and summer (SUM).
Supplementary Figure 10 | Relative transcript abundance (TPM) of nitrogen metabolism-related genes. Color bars represent the relative contribution of main taxonomic groups to the transcription of each gene at the costal and offshore station during winter (WIN), spring (SPG), and summer (SUM).
Supplementary Figure 11 | Relative transcript abundance (TPM) of phosphorus metabolism-related genes. Color bars represent the relative contribution of main taxonomic groups to the transcription of each gene at the costal and offshore station during winter (WIN), spring (SPG), and summer (SUM).
Supplementary Figure 12 | Relative transcript abundance (TPM) of sulfur metabolism-related genes. Color bars represent the relative contribution of main taxonomic groups to the transcription of each gene at the costal and offshore station during winter (WIN), spring (SPG), and summer (SUM).
Supplementary Figure 13 | Relative transcript abundance (TPM) of iron metabolism-related genes. Color bars represent the relative contribution of main taxonomic groups to the transcription of each gene at the costal and offshore station during winter (WIN), spring (SPG), and summer (SUM).
Supplementary Figure 14 | Relative transcript abundance (TPM) of vitamin-related genes. Color bars represent the relative contribution of main taxonomic groups to the transcription of each gene at the costal and offshore station during winter (WIN), spring (SPG), and summer (SUM).
Supplementary Figure 15 | Relative transcript abundance (TPM) of response to stress-related genes. Color bars represent the relative contribution of main taxonomic groups to the transcription of each gene at the costal and offshore station during winter (WIN), spring (SPG), and summer (SUM).
Supplementary Figure 16 | Relative transcript abundance (TPM) of response to stimuli-related genes. Color bars represent the relative contribution of main taxonomic groups to the transcription of each gene at the costal and offshore station during winter (WIN), spring (SPG), and summer (SUM).
Supplementary Figure 17 | Relative transcript abundance (TPM) of phototrophy-related genes. Color bars represent the relative contribution of main taxonomic groups to the transcription of each gene at the costal and offshore station during winter (WIN), spring (SPG), and summer (SUM).
Supplementary Figure 18 | Daily variability of carbon-, nitrogen- and phosphorus-related genes at the costal and offshore stations during winter (WIN), spring (SPG), and summer (SUM). Relative transcript abundance (TPM) of selected key biological process is provided. Color bars represent the relative contribution of main taxonomic groups to transcription. The value for each sampling day (days 1, 3, 5 and 7) is shown. Note that different scales are used on the y-axis.
Supplementary Figure 19 | Daily variability of sulfur-, iron- and vitamins-related genes at the costal and offshore stations during winter (WIN), spring (SPG), and summer (SUM). Relative transcript abundance (TPM) of selected key biological process is provided. Color bars represent the relative contribution of main taxonomic groups to transcription. The value for each sampling day (days 1, 3, 5 and 7) is shown. Note that different scales are used on the y-axis.
Supplementary Figure 20 | Daily variability of response to stimuli-, response to stress- and phototrophy-related genes at the costal and offshore stations during winter (WIN), spring (SPG), and summer (SUM). Relative transcript abundance (TPM) of selected key biological process is provided. Color bars represent the relative contribution of main taxonomic groups to transcription. The value for each sampling day (days 1, 3, 5 and 7) is shown. Note that different scales are used on the y-axis.
Supplementary Figure 21 | Daily variability (estimated as the daily TPMs of each gene divided by the mean value of the TPMs of that gene during the four days of the specific season and station) of selected genes in the carbon, nitrogen and phosphorus metabolism categories at the offshore and costal stations during winter (WIN), spring (SPG), and summer (SUM). The color and the vertical position of the bubble (y-axis) represent the value of the ratio. The size of the bubble represents the daily relative abundance (TPM) of each gene over the days of sampling (days 1, 3, 5, and 7; x-axis).
Supplementary Figure 22 | Daily variability (estimated as the daily TPMs of each gene divided by the mean value of the TPMs of that gene during the four days of the specific season and station) of selected genes in the sulfur, iron and vitamins metabolism categories at the offshore and costal stations during winter (WIN), spring (SPG), and summer (SUM). The color and the vertical position of the bubble (y-axis) represent the value of the ratio. The size of the bubble represents the daily relative abundance (TPM) of each gene over the days of sampling (days 1, 3, 5, and 7; x-axis).
Supplementary Figure 23 | Daily variability (estimated as the daily TPMs of each gene divided by the mean value of the TPMs of that gene during the four days of the specific season and station) of selected genes in the response to stimuli, response to stress, and phototrophy categories at the offshore and costal stations during winter (WIN), spring (SPG), and summer (SUM). The color and the vertical position of the bubble (y-axis) represent the value of the ratio. The size of the bubble represents the daily relative abundance (TPM) of each gene over the days of sampling (days 1, 3, 5, and 7; x-axis).
References
Ahlgren N. A., Belisle B. S., Lee M. D. (2020). Genomic mosaicism underlies the adaptation of marine Synechococcus ecotypes to distinct oceanic iron niches. Environ. Microbiol. 22, 1801–1815. doi: 10.1111/1462-2920.14893
Alonso-Sáez L., Morán X. A. G., González J. M. (2020). Transcriptional patterns of biogeochemically relevant marker genes by temperate marine bacteria. Front. Microbiol. 11, 1–10. doi: 10.3389/fmicb.2020.00465
Álvarez-Salgado X. A., Beloso S., Joint I., Nogueira E., Chou L., Pérez F. F., et al. (2002). New production of the NW Iberian shelf during the upwelling season over the period 1982-1999. Deep Res. Part I Oceanogr Res. Pap 49, 1725–1739. doi: 10.1016/S0967-0637(02)00094-8
Amin S. A., Green D. H., Hart M. C., Kü F. C., Sunda W. G., Carrano C. J., et al. (2009). Photolysis of iron-siderophore chelates promotes bacterial-algal mutualism. Proceedings of the National Academy of Sciences 106 (40), 17071–17076. doi: 10.1073/pnas.0905512106
Amin S. A., Hmelo L. R., Van Tol H. M., Durham B. P., Carlson L. T., Heal K. R., et al. (2015). Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522, 98–101. doi: 10.1038/nature14488
Archer S. D., Cummings D. G., Llewellyn C. A., Fishwick J. R. (2009). Phytoplankton taxa, irradiance and nutrient availability determine the seasonal cycle of DMSP in temperate shelf seas. Mar. Ecol. Prog. Ser. 394, 111–124. doi: 10.3354/meps08284
Arístegui J., Barton E. D., Álvarez-Salgado X. A., Santos A. M. P., Figueiras F. G., Kifani S., et al. (2009). Sub-regional ecosystem variability in the Canary Current upwelling. Prog. Oceanogr 83, 33–48. doi: 10.1016/j.pocean.2009.07.031
Barbosa A. B., Galvão H. M., Mendes P. A., Álvarez-Salgado X. A., Figueiras F. G., Joint I. (2001). Short-term variability of heterotrophic bacterioplankton during upwelling off the NW Iberian margin. Prog. Oceanogr 51, 339–359. doi: 10.1016/S0079-6611(01)00074-X
Bergen B., Herlemann D. P. R., Jürgens K. (2015). Zonation of bacterioplankton communities along aging upwelled water in the northern Benguela upwelling. Front. Microbiol. 6, 1–12. doi: 10.3389/fmicb.2015.00621
Bertrand E. M., Allen A. E. (2012). Influence of vitamin B auxotrophy on nitrogen metabolism in eukaryotic phytoplankton. Front. Microbiol. 3, 1–16. doi: 10.3389/fmicb.2012.00375
Blanton J. O., Atkinson L. P., De Castillejo F. F., Montero A. L. (1984). Coastal upwelling off the Rias Bajas, Galicia, northwest Spain I: Hydrographic studies. Coast. upwelling off Rias Bajas Galicia Northwest Spain II Benthic Stud. 183, 79–90.
Buchfink B., Xie C., Huson D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Bunse C., Israelsson S., Baltar F., Bertos-Fortis M., Fridolfsson E., Legrand C., et al. (2019). High frequency multi-year variability in baltic sea microbial plankton stocks and activities. Front. Microbiol. 10, 1–18. doi: 10.3389/fmicb.2018.03296
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caron D. A. (1994). Inorganic nutrients, bacteria, and the microbial loop. Microb. Ecol. 28, 295–298. doi: 10.1007/BF00166820
Chénard C., Wijaya W., Vaulot D., Lopes dos Santos A., Martin P., Kaur A., et al. (2019). Temporal and spatial dynamics of Bacteria, Archaea and protists in equatorial coastal waters. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-019-52648-x
Costas-Selas C., Martínez-García S., Logares R., Hernández-Ruiz M., Teira E. (2023). Role of bacterial community composition as a driver of the small-sized phytoplankton community structure in a productive coastal system. Microb. Ecol. 86(2), 777–794. doi: 10.1007/s00248-022-02125-2
Croft M. T., Lawrence A. D., Raux-Deery E., Warren M. J., Smith A. G. (2005). Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 438, 90–93. doi: 10.1038/nature04056
Cury J. C., Araujo F. V., Coelho-Souza S. A., Peixoto R. S., Oliveira J. A. L., Santos H. F., et al. (2011). Microbial diversity of a Brazilian coastal region influenced by an upwelling system and anthropogenic activity. PloS One 6 (1), e16553. doi: 10.1371/journal.pone.0016553
De Corte D., Muck S., Tiroch J., Mena C., Herndl G. J., Sintes E. (2021). Microbes mediating the sulfur cycle in the Atlantic Ocean and their link to chemolithoautotrophy. Environ. Microbiol. 00, 1–16. doi: 10.1111/1462-2920.15759
Del Fabbro C., Scalabrin S., Morgante M., Giorgi F. M. (2013). An extensive evaluation of read trimming effects on illumina NGS data analysis. PloS One 8, 1–13. doi: 10.1371/journal.pone.0085024
Durham B. P., Boysen A. K., Carlson L. T., Groussman R. D., Heal K. R., Cain K. R., et al. (2019). Sulfonate-based networks between eukaryotic phytoplankton and heterotrophic bacteria in the surface ocean. Nat. Microbiol. 4, 1706–1715. doi: 10.1038/s41564-019-0507-5
Dyhrman S. T., Ammerman J. W., van Mooy B. A. S. (2007). Microbes and the marine phosphorus cycle. Oceanography 20, 110–116. doi: 10.5670/oceanog.2007.54
Ewels P., Magnusson M., Lundin S., Käller M. (2016). MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. doi: 10.1093/bioinformatics/btw354
Fernández-Gómez B., Richter M., Schüler M., Pinhassi J., Acinas S. G., González J. M., et al. (2013). Ecology of marine bacteroidetes: A comparative genomics approach. ISME J. 7, 1026–1037. doi: 10.1038/ismej.2012.169
Figueiras F. G., Labarta U., Fernández Reiriz M. J. (2002). Coastal upwelling, primary production and mussel growth in the Rías Baixas of Galicia. Hydrobiologia 484, 121–131. doi: 10.1023/A:1021309222459
Figueiras F. G., Ríos A. F. (1993). Phytoplankton Succession, Red Tides and the Hydrographic Regime in the Rias Bajas of Galicia. Dev. Mar. Biol 1993.
Forchhammer K., Selim K. A., Huergo L. F. (2022). New views on PII signaling: from nitrogen sensing to global metabolic control. Trends Microbiol. 30, 722–735. doi: 10.1016/j.tim.2021.12.014
Gasol J. M., Del Giorgio P. A. (2000). Using flow cytometry for counting natural planktonic bacteria and understanding the structure of planktonic bacterial communities. Sci. Mar. 64, 197–224. doi: 10.3989/scimar.2000.64n2197
Grossman A. (2016). Nutrient acquisition: the generation of bioactive vitamin B12 by microalgae. Curr. Biol. 26, R319–R321. doi: 10.1016/j.cub.2016.02.047
Guillou L., Bachar D., Audic S., Bass D., Berney C., Bittner L., et al. (2012). The Protist Ribosomal Reference database (PR2): A catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41 (D1), D597–D604. doi: 10.1093/nar/gks1160
Harke M. J., Gobler C. J. (2013). Global transcriptional responses of the toxic cyanobacterium, microcystis aeruginosa, to nitrogen stress, phosphorus stress, and growth on organic matter. PloS One 8 (7), e69834. doi: 10.1371/journal.pone.0069834
Hernández-Ruiz M., Prieto A., Barber-Lluch E., Teira E. (2018). Amino acid utilization by eukaryotic picophytoplankton in a coastal upwelling system. Mar. Ecol. Prog. Ser. 588, 43–57. doi: 10.3354/meps12435
Huson D. H., Beier S., Flade I., Górska A., El-Hadidi M., Mitra S., et al. (2016). MEGAN community edition - interactive exploration and analysis of large-scale microbiome sequencing data. PloS Comput. Biol. 12, 1–12. doi: 10.1371/journal.pcbi.1004957
Jimenez-Infante F., Ngugi D. K., Vinu M., Blom J., Alam I., Bajic V. B., et al. (2017). Genomic characterization of two novel SAR11 isolates from the Red Sea, including the first strain of the SAR11 Ib clade. FEMS Microbiol. Ecol. 93, 1–10. doi: 10.1093/femsec/fix083
Joglar V., Álvarez-Salgado X. A., Gago-Martinez A., Leao J. M., Pérez-Martínez C., Pontiller B., et al. (2021a). Cobalamin and microbial plankton dynamics along a coastal to offshore transect in the Eastern North Atlantic Ocean. Environ. Microbiol. 23, 1559–1583. doi: 10.1111/1462-2920.15367
Joglar V., Pontiller B., Martínez-García S., Fuentes-Lema A., Pérez-Lorenzo M., Lundin D., et al. (2021b). Microbial plankton community structure and function system. Appl. Env. Microbiol. 87 (22), e01525–21. doi: 10.1128/AEM.01525-21
Joglar V., Prieto A., Barber-Lluch E., Hernández-Ruiz M., Fernández E., Teira E. (2020). Spatial and temporal variability in the response of phytoplankton and bacterioplankton to B-vitamin amendments in an upwelling system. Biogeosciences Discuss 17(10), 2807–2823. doi: 10.5194/bg-2019-306
Joshi N., Sickle F. J. N. (2011). a sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33)[Software].
Kämpf J., Chapman P. (2016). Upwelling systems of the world. (Switzerland: Springer International Publishing), 31–42.
Kerkhof L. J., Voytek M. A., Sherrell R. M., Millie D., Schofield O. (1999). Variability in bacterial community structure during upwelling in the coastal ocean. Hydrobiologia 401, 139–148. doi: 10.1007/978-94-011-4201-4_11
Kiene R. P., Linn L. J., Bruton J. A. (2000). New and important roles for DMSP in marine microbial communities. J. Sea Res. 43, 209–224. doi: 10.1016/S1385-1101(00)00023-X
Klindworth A., Mann A. J., Huang S., Wichels A., Quast C., Waldmann J., et al. (2014). Diversity and activity of marine bacterioplankton during a diatom bloom in the North Sea assessed by total RNA and pyrotag sequencing. Mar. Genomics 18, 185–192. doi: 10.1016/j.margen.2014.08.007
Koch F. (2012). The Role of Vitamin B 1 and B 12 in Controlling Phytoplankton Biomass, Diversity, and Dynamics. (Doctoral dissertation, State University of New York at Stony Brook).
Koch F., Trimborn S. (2019). Limitation by fe, zn, co, and b12 results in similar physiological responses in two antarctic phytoplankton species. Front. Mar. Sci. 6. doi: 10.3389/fmars.2019.00514
Lin Q., Shang L., Wang X., Hu Z., Du H., Wang H. (2021). Different dimethylsulphoniopropionate-producing ability of dinoflagellates could affect the structure of their associated bacterial community. Algal Res. 57, 102359. doi: 10.1016/j.algal.2021.102359
Liu Y., Blain S., Crispi O., Rembauville M., Obernosterer I. (2020). Seasonal dynamics of prokaryotes and their associations with diatoms in the Southern Ocean as revealed by an autonomous sampler. Environ. Microbiol. 22, 3968–3984. doi: 10.1111/1462-2920.15184
Liu Z., Jones A. C., Campbell V., Hambright K. D., Heidelberg K. B., Caron D. A. (2015). Gene expression in the mixotrophic prymnesiophyte, Prymnesium parvum, responds to prey availability. Front. Microbiol. 6, 1–12. doi: 10.3389/fmicb.2015.00319
Logares R., Audic S., Bass D., Bittner L., Boutte C., Christen R., et al. (2014). Patterns of rare and abundant marine microbial eukaryotes. Curr. Biol. 24, 813–821. doi: 10.1016/j.cub.2014.02.050
Lorenzen C. J., Newton Downs J. (1986). The specific absorption coefficients of chlorophyllide a and pheophorbide a in 90% acetone, and comments on the fluorometric determination of chlorophyll and pheopigments. Limnol Oceanogr 31, 449–452.
Marcel M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12.
Martínez-García S., Fernández E., Álvarez-Salgado X. A., González J., Lønborg C., Marañón E., et al. (2010). Differential responses of phytoplankton and heterotrophic bacteria to organic and inorganic nutrient additions in coastal waters off the NW Iberian Peninsula. Mar. Ecol. Prog. Ser. 416, 17–33. doi: 10.3354/meps08776
Massana R., Gobet A., Audic S., Bass D., Bittner L., Boutte C., et al. (2015). Marine protist diversity in European coastal waters and sediments as revealed by high-throughput sequencing. Environ. Microbiol. 17, 4035–4049. doi: 10.1111/1462-2920.12955
Moran M. A., González J. M., Kiene R. P. (2003). Linking a bacterial taxon to sulfur cycling in the sea: Studies of the marine Roseobacter group. Geomicrobiol J. 20, 375–388. doi: 10.1080/01490450303901
Muratore D., Boysen A. K., Harke M. J., Becker K. W., Casey J. R., Coesel S. N., et al. (2022). Complex marine microbial communities partition metabolism of scarce resources over the diel cycle. Nat. Ecol. Evol. 6, 218–229. doi: 10.1038/s41559-021-01606-w
Needham D. M., Sachdeva R., Fuhrman J. A. (2017). Ecological dynamics and co-occurrence among marine phytoplankton, bacteria and myoviruses shows microdiversity matters. ISME J. 11, 1614–1629. doi: 10.1038/ismej.2017.29
Nogueira E., Pérez F. F., Ríos A. F. (1997). Seasonal patterns and long-term trends in an estuarine upwelling ecosystem (Ria de Vigo, NW Spain). Estuar. Coast. Shelf Sci. 44, 285–300. doi: 10.1006/ecss.1996.0119
Nowinski B., Moran M. A. (2021). Niche dimensions of a marine bacterium are identified using invasion studies in coastal seawater. Nat. Microbiol. 6, 524–532. doi: 10.1038/s41564-020-00851-2
O’Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., McVeigh R., et al. (2016). Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Olson D. K., Yoshizawa S., Boeuf D., Iwasaki W., Delong E. F. (2018). Proteorhodopsin variability and distribution in the North Pacific Subtropical Gyre. ISME J. 12, 1047–1060. doi: 10.1038/s41396-018-0074-4
Ottesen E. A., Young C. R., Eppley J. M., Ryan J. P., Chavez F. P., Scholin C. A., et al. (2013). Pattern and synchrony of gene expression among sympatric marine microbial populations. Proc. Natl. Acad. Sci. U.S.A. 110 (6), E488–E497. doi: 10.1073/pnas.1222099110
Ottesen E. A., Young C. R., Gifford S. M., Eppley J. M., Marin R., Schuster S. C., et al. (2014). Multispecies diel transcriptional oscillations in open ocean heterotrophic bacterial assemblages. Science 345, 207–212. doi: 10.1126/science.1252476
Pajares S., Varona-Cordero F., Hernández-Becerril D. U. (2020). Spatial distribution patterns of bacterioplankton in the oxygen minimum zone of the tropical mexican pacific. Microb. Ecol. 80, 519–536. doi: 10.1007/s00248-020-01508-7
Parada A. E., Needham D. M., Fuhrman J. A. (2016). Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Pinhassi J., DeLong E. F., Béjà O., González J. M., Pedrós-Alió C. (2016). Marine bacterial and archaeal ion-pumping rhodopsins: genetic diversity, physiology, and ecology. Microbiol. Mol. Biol. Rev. 80, 929–954. doi: 10.1128/MMBR.00003-16
Pinhassi J., Sala M. M., Havskum H., Peters F., Guadayol Ò., Malits A., et al. (2004). Changes in bacterioplankton composition under different phytoplankton regimens. Appl. Environ. Microbiol. 70, 6753–6766. doi: 10.1128/AEM.70.11.6753-6766.2004
Pisareva M. N., Pickart R. S., Lin P., Fratantoni P. S., Weingartner T. J. (2019). On the nature of wind-forced upwelling in Barrow Canyon. Deep Res. Part II Top. Stud. Oceanogr 162, 63–78. doi: 10.1016/j.dsr2.2019.02.002
Pontiller B., Martínez-García S., Joglar V., Amnebrink D., Pérez-Martínez C., González J. M., et al. (2022). Rapid bacterioplankton transcription cascades regulate organic matter utilization during phytoplankton bloom progression in a coastal upwelling system. ISME J. 16, 2360–2372. doi: 10.1038/s41396-022-01273-0
Pontiller B., Pérez-martínez C., Bunse C., Osbeck C. M. G. (2021). Taxon-specific shifts in bacterial and archaeal transcription of dissolved organic matter cycling genes in a stratified fjord. Msystems. 6 (6), e00575-21. doi: 10.1128/mSystems.00575-21
Poretsky R. S., Sun S., Mou X., Moran M. A. (2010). Transporter genes expressed by coastal bacterioplankton in response to dissolved organic carbon. Environ. Microbiol. 12, 616–627. doi: 10.1111/j.1462-2920.2009.02102.x
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Reji L., Tolar B. B., Chavez F. P., Francis C. A. (2020). Depth-differentiation and seasonality of planktonic microbial assemblages in the monterey bay upwelling system. Front. Microbiol. 11, 1–14. doi: 10.3389/fmicb.2020.01075
Rivkin R. B., Legendre L. (2001). Biogenic carbon cycling in the upper ocean: Effects of microbial respiration. Science 291, 2398–2400. doi: 10.1126/science.291.5512.2398
Robidart J. C., Magasin J. D., Shilova I. N., Turk-Kubo K. A., Wilson S. T., Karl D. M., et al. (2019). Effects of nutrient enrichment on surface microbial community gene expression in the oligotrophic North Pacific Subtropical Gyre. ISME J. 13, 374–387. doi: 10.1038/s41396-018-0280-0
Ruiz-Perez C. A., Bertagnolli A. D., Tsementzi D., Woyke T., Stewart F. J., Konstantinidis K. T. (2021). Description of Candidatus Mesopelagibacter carboxydoxydans and Candidatus Anoxipelagibacter denitrificans: Nitrate-reducing SAR11 genera that dominate mesopelagic and anoxic marine zones. Syst. Appl. Microbiol. 44, 126185. doi: 10.1016/j.syapm.2021.126185
Sañudo-Wilhelmy S. A., Cutter L. S., Durazo R., Smail E. A., Gómez-Consarnau L., Webb E. A., et al. (2012). Multiple B-vitamin depletion in large areas of the. Proc. Natl. Acad. Sci. U.S.A. 109, 14041–14045. doi: 10.1073/pnas.1208755109
Satinsky B. M., Crump B. C., Smith C. B., Sharma S., Zielinski B. L., Doherty M., et al. (2014). Microspatial gene expression patterns in the Amazon River Plume. Proc. Natl. Acad. Sci. U.S.A. 111, 11085–11090. doi: 10.1073/pnas.1402782111
Satinsky B. M., Smith C. B., Sharma S., Landa M., Medeiros P. M., Coles V. J., et al. (2017). Expression patterns of elemental cycling genes in the Amazon River Plume. ISME J. 11, 1852–1864. doi: 10.1038/ismej.2017.46
Shilova I. N., Robidart J. C., James Tripp H., Turk-Kubo K., Wawrik B., Post A. F., et al. (2014). A microarray for assessing transcription from pelagic marine microbial taxa. ISME J. 8, 1476–1491. doi: 10.1038/ismej.2014.1
Smriga S., Fernandez V. I., Mitchell J. G., Stocker R. (2016). Chemotaxis toward phytoplankton drives organic matter partitioning among marine bacteria. Proc. Natl. Acad. Sci. U.S.A. 113, 1576–1581. doi: 10.1073/pnas.1512307113
Souto C., Fariña-Busto L., Alvarez E., Rodríguez I. (2001). Wind and tide current prediction using a 3D finite difference model in the Ría de Vigo (NW Spain). Sci. Mar. 65, 269–276. doi: 10.3989/scimar.2001.65s1269
Stock C. A., John J. G., Rykaczewski R. R., Asch R. G., Cheung W. W. L., Dunne J. P., et al. (2017). Reconciling fisheries catch and ocean productivity. Proc. Natl. Acad. Sci. U.S.A. 114, E1441–E1449. doi: 10.1073/pnas.1610238114
Stoecker D. K., Hansen P. J., Caron D. A., Mitra A. (2017). Mixotrophy in the marine plankton. Ann. Rev. Mar. Sci. 9, 311–335. doi: 10.1146/annurev-marine-010816-060617
Teeling H., Fuchs B. M., Becher D., Klockow C., Gardebrecht A., Bennke C. M., et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611. doi: 10.1126/science.1218344
Teeling H., Fuchs B. M., Bennke C. M., Krüger K., Chafee M., Kappelmann L., et al. (2016). Recurring patterns in bacterioplankton dynamics during coastal spring algae blooms. Elife 5, 1–31. doi: 10.7554/eLife.11888
Teira E., Hernández-Ruiz M., Barber-Lluch E., Sobrino C., Teixeira I. G., Álvarez-Salgado X. A., et al. (2016). Bacterioplankton responses to riverine and atmospheric inputs in a coastal upwelling system (Ría de Vigo, NW Spain). Mar. Ecol. Prog. Ser. 542, 39–50. doi: 10.3354/meps11565
Teira E., Hernando-Morales V., Fernández A., Martínez-García S., Álvarez-Salgado X. A., Bode A., et al. (2015). Local differences in phytoplankton-bacterioplankton coupling in the coastal upwelling off Galicia (NW Spain). Mar. Ecol. Prog. Ser. 528, 53–69. doi: 10.3354/meps11228
Teixeira I. G., Arbones B., Froján M., Nieto-cid M., Álvarez-Salgado X. A., Castro C. G., et al. (2018). Response of phytoplankton to enhanced atmospheric and riverine nutrient inputs in a coastal upwelling embayment. Estuar. Coast. Shelf Sci. 210, 132–141. doi: 10.1016/j.ecss.2018.06.005
Varaljay V. A., Gifford S. M., Wilson S. T., Sharma S., Karl D. M., Moran M. A. (2012). Bacterial dimethylsulfoniopropionate degradation genes in the oligotrophic North Pacific subtropical gyre. Appl. Environ. Microbiol. 78, 2775–2782. doi: 10.1128/AEM.07559-11
Wang M., Wang H., Wang P., Fu H. H., Li C. Y., Qin Q. L., et al. (2022). TCA cycle enhancement and uptake of monomeric substrates support growth of marine Roseobacter at low temperature. Commun. Biol. 5 (1), 705. doi: 10.1038/s42003-022-03631-2
Wear E. K., Carlson C. A., James A. K., Brzezinski M. A., Windecker L. A., Nelson C. E. (2015). Synchronous shifts in dissolved organic carbon bioavailability and bacterial community responses over the course of an upwelling-driven phytoplankton bloom. Limnology and Oceanography 60 (2), 657–677. doi: 10.1002/lno.10042
Welschmeyer N. A. (1994). Fluorometric analysis of chlorophyll a in the presence of chlorophyll b and pheopigments. Limnol Oceanogr 39, 1985–1992. doi: 10.4319/lo.1994.39.8.1985
Zdanowski M. K., Figueiras F. G. (1997). Relationships between the abundance of bacteria and other biota and the hydrographic variability in the Ria de Vigo, Spain. Mar. Ecol. Prog. Ser. 147, 257–267. doi: 10.3354/meps147257
Zhang J., Kobert K., Flouri T., Stamatakis A. (2014). PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. doi: 10.1093/bioinformatics/btt593
Zhao Y., Schlundt C., Booge D., Bange H. W. (2021). A decade of dimethyl sulfide (DMS), dimethylsulfoniopropionate (DMSP) and dimethyl sulfoxide (DMSO) measurements in the southwestern Baltic Sea. Biogeosciences 18, 2161–2179. doi: 10.5194/bg-18-2161-2021
Keywords: bacterioplankton, upwelling systems, phytoplankton bloom, metatranscriptomics, metabarcoding
Citation: Delgadillo-Nuño E, Teira E, Pontiller B, Lundin D, Joglar V, Pedrós-Alió C, Fernández E, Pinhassi J and Martínez-García S (2024) Coastal upwelling systems as dynamic mosaics of bacterioplankton functional specialization. Front. Mar. Sci. 10:1259783. doi: 10.3389/fmars.2023.1259783
Received: 26 July 2023; Accepted: 27 November 2023;
Published: 04 January 2024.
Edited by:
Jin Zhou, Tsinghua University, ChinaReviewed by:
Daniel Jonathan Sher, University of Haifa, IsraelFabiano Thompson, Federal University of Rio de Janeiro, Brazil
Copyright © 2024 Delgadillo-Nuño, Teira, Pontiller, Lundin, Joglar, Pedrós-Alió, Fernández, Pinhassi and Martínez-García. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Martínez-García, sandra@uvigo.gal