
Abstract
Saline, 450-m-deep Lake Van (Eastern Anatolia, Turkey) is, with 576 km3, the third largest closed lake on Earth and its largest soda lake. In 1989 and 1990, we investigated the hydrochemistry of the lake’s water column and of the tributary rivers. We also cored the Postglacial sediment column at various water depths. The sediment is varved throughout, allowing precise dating back to ca. 15 ka BP. Furthermore, lake terrace sediments provided a 606-year-long floating chronology of the Glacial high-stand of the lake dating to 21 cal. ka BP. The sediments were investigated for their general mineralogical composition, important geochemical parameters, and pore water chemistry as well. These data allow reconstructing the history of the lake level that has seen several regressions and transgressions since the high-stand at the end of the Last Glacial Maximum. Today, the lake is very alkaline, highly supersaturated with Ca-carbonate and has a salt content of about 22 g kg−1. In summer, the warmer epilimnion is diluted with river water and forms a stable surface layer. Depth of winter mixing differs from year to year but during time of investigation the lake was oxygenated down to its bottom. In general, the lake is characterized by an Na–CO3–Cl–(SO4)-chemistry that evolved from the continuous loss of calcium as carbonate and magnesium in the form of Mg-silica-rich mineral phases. The Mg cycle is closely related to that of silica which in turn is governed by the production and dissolution of diatoms as the dominant phytoplankton species in Lake Van. In addition to Ca and Mg, a mass balance approach based on the recent lake chemistry and river influx suggests a fractional loss of potassium, sodium, sulfur, and carbon in comparison to chloride in the compositional history of Lake Van. Within the last 3 ka, minor lake level changes seem to control the frequency of deep water renewal, the depth of stratification, and the redox state of the hypolimnion. Former major regressions are marked by Mg-carbonate occurrences in the otherwise Ca-carbonate dominated sediment record. Pore water data suggest that, subsequent to the major regression culminating at 10.7 ka BP, a brine layer formed in the deep basin that existed for about 7 ka. Final overturn of the lake, triggered by the last major regression starting at about 3.5 ka BP, may partly account for the relative depletion in sulfur and carbon due to rapid loss of accumulated gases. An even stronger desiccation phase is proposed for the time span between about 20 and 15 ka BP following the LGM, during which major salts could have been lost by precipitation of Na-carbonates and Na-sulfates.
Similar content being viewed by others

1 Introduction
The sediment records of deep lakes give researchers the opportunity to study climatic fluctuations of the recent geological past. The kind and amount of sediments deposited is governed by the size and geology of the basin and by the lake’s water balance, i.e., the general climate of the region in terms of temperature, precipitation, and evaporation. Geology and climate determine the dissolved and detrital load of tributary rivers, the water chemistry of the lake and its temperature, as well as the type of lake sediment. Terminal deep lakes are particularly suitable for paleoclimate studies because (i) their water level reacts very sensitively to climate changes (e.g., Langbein 1961) and (ii) all materials brought in by rivers or via atmospheric transport are deposited within the lake basin making mass budget calculations simpler.
Lake Van, or Van Gölü in Turkish, is located in the highlands of eastern Anatolia, Turkey, at 38° 38′ N, 42° 49′ E. Its large size (3,522 km2), its high altitude (1648 m a.s.l.), its pronounced water level changes and its peculiar alkaline waters attracted many early travelers, among them Parrot (1834), Abich (1856), and Sieger (1888). By volume (576 km3) Lake Van is the third largest terminal lake on Earth (Fig. 1) after the Caspian Sea and Lake Issyk-Kul. The lake has a maximum depth of 451 m, ranking it among the deepest lakes worldwide. Lake Van is also the largest soda lake on Earth, and served as a model of the possible highly alkaline chemistry of the early ocean (e.g., Kempe and Degens 1985; Kempe and Kaźmierczak 1994, 2002, 2003, 2007). Extensive information about the hydrography, hydrochemistry, and geochemistry of lake sediments was obtained during a multidisciplinary study carried out in 1974 (Kempe 1977; Degens and Kurtman 1978; Degens et al. 1984). During the expedition the depth of the lake was measured for the first time and the first bathymetric map was constructed (Wong and Degens 1978). The survey with a 3.5-kHz profiling system (Wong et al. 1978) exposed a karst-like topography between depths of 100 and 200 m on the eastern shelf interpreted as erosion channels build during a lake level low stand. Subaerial downcutting of rivers left behind relic deltas 40–60 km away from the present river mouths (Degens et al. 1984). Air gun profiles showed the presence of up to 600 m of unconsolidated sediments, composed of extensive, well-stratified layers alternating with chaotically reflecting layers. The stratified sections were deposited during periods of normal sedimentation, while the chaotic layers are thought to represent slumps deposited during rapid changes in lake level that caused sediment instability and hence slumping (Wong and Finckh 1978).

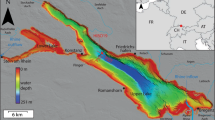
Situation of Lake Van, its bathymetry and drainage area. Positions of core stations in 1974 and 1990, positions of lake water profiling and river water sampling locations as given in the legend. Lettering refers to core stations discussed in the text. (Bathymetry according to the sea chart of the Directorate for Hydrography and Oceanography 1985)
The hydrochemistry of the lake and of all rivers feeding Lake Van was investigated in great detail in 1989 and 1990 (Reimer 1995). Additionally, the microbialites were surveyed and sampled (Kempe et al. 1991; Kaźmierczak and Kempe 2003; López-García et al. 2005) and the physiology of the Lake Van endemic fish Chalcalburnus tarichi (Cyprinidae) was investigated (Danulat and Kempe 1992).
Up to 10-m-long sediment cores were recovered from the deep basin of the lake, covering a time period of about 15 ka (Figs. 1 and 2) (Landmann 1996; Lemcke 1996). The results of the Lake Van sediment investigations are published elsewhere (see Appendix), but their interpretation is summarized below. Here, we present chemical analyses of lake-, river-, and downcore trends of pore waters. These data allow mass balance calculations and provide information about the carbonate system, diagenetic processes and the mixing behavior of Lake Van yielding clues as to the history of the lake level and the lake chemistry in the Postglacial.

Lake level fluctuations of Lake Van in the past 20 ka and sediment porosity versus age. Shaded areas mark periods with negative water balance. Maximum depth of Lake Van today is 451 m, water depths of the cores are given in the legend. Note that the scale of the salinity axis is not linear
2 Material and Methods
The Lake Van expeditions of 1989 and 1990 (both in June/July) used the tug boat “Süphan” out of Tatvan harbor as a “research vessel” to run echosounding profiles, sample the water column, collect sediment cores and explore by diving the up to 40-m-tall microbialites on the lake bottom (Kempe et al. 1991). Most analyses were carried out in a field lab set up in an old office of the Turkish Railway.
In both years, water samples were collected from the water column in the central lake basin (Tatvan Basin) and in near-shore mixing zones at the mouth of tributaries. Hydrochemical samples were also taken from major and minor tributaries, lakes and springs in the vicinity of Lake Van, and at sublacustrine springs by diving. In case of the depth profiling, sampling was conducted using 10 l Niskin bottles, otherwise samples were directly filled into PE-bottles. Temperature, electrical conductivity, pH, and redox potential were recorded in situ where possible or directly following the sampling. Measurements were conducted with handheld instruments (WTW, Germany), and pH electrodes (Schott, Germany) were calibrated against NBS buffers (Ingold, Germany). Water samples were filtered through Whatman GFF glass fiber filters and Nucleopore polycarbonate filters to determine the particulate fraction. Filtered samples were fixed by acidification or adding HgCl2 for later analyses.
In 1990, sediment cores were retrieved at ten stations using a Kullenberg-type piston corer, with the longest core recovered measuring 9 m. For upright transport, sediment cores were cut into 1-m-long sections. Core segments were partly opened in the field laboratory in Tatvan or one to two months later in the laboratory at the University of Hamburg for geochemical subsampling. Electrical conductivity, pH, and redox potential of pore water were recorded immediately after squeezing subsamples under nitrogen atmosphere in a pressure device. Water was then filtrated and preserved for further analyses. Pore water pH was measured at laboratory temperature of 20 ± 3°C. For mineral saturation calculations, pH at in situ temperature (i.e., 3.3°C at lake bottom) was approximated using the equation from Millero (1979).
Total alkalinity was determined by an automated potentiometric titration system (Metrohm 665 motor burette and Schott CG 841 pH-meter) immediately after sample recovery. Titrations were performed at 20°C using a sample volume of 100 ml and pre-adjusted HCl as titrant (1 N ± 0.1%, Merck, Germany) for lake water samples. The same volume but pre-adjusted 0.1 N ± 0.1% HCl (Merck, Germany) was used for river water samples while aliquots of 500 μl and 0.01 N HCl were used in case of pore water samples. Dissolved oxygen was analyzed titrimetrically following the Winkler method. Nutrients (PO4, NH4, NO2, NO3, and dissolved silica) were analyzed spectrophotometrically using slightly modified methods given in Grasshoff et al. (1983).
Main cations (Na, K, Mg, and Ca) of the lake water were determined by flame AES (atomic emission spectroscopy) or flame AAS (atomic absorption spectroscopy) at less sensitive wavelengths (Perkin Elmer). Additionally, ICP-OES (Perkin Elmer Plasma II) was used to determine main cations as well as Sr, Ba, and B in river and spring fresh waters.
Ion chromatography with indirect photometric detection was applied to measure chloride and sulfate concentrations as well as nitrate concentrations in river and spring waters. Chloride concentrations were also measured by photometric analysis.
In case of tributary waters, iron and manganese were measured by flameless AAS, while TXRFA (total reflection X-ray fluorescence analysis, GKSS, Geesthacht, Germany) was applied on samples from Lake Van depth profiles.
Total carbon and total nitrogen determination on sediment samples was conducted with a Carlo-Erba CN-analyzer 1500, and organic carbon contents were analyzed with a LECO CS-analyzer after removing the carbonate content by acidification. Carbonate carbon was calculated as the difference of total and organic carbon, and as a control, in part directly determined using a Carmograph 6 carbonate analyzer (Wösthoff, Germany). Amounts of particulate phosphorus, both the organically and inorganically bound fractions, and of opaline silica in the sediments were photometrically measured after varying digestion techniques following the methods given in Liebezeit (1991) and Mortlock and Froelich (1989).
3 The Lake Van Sediment Record
In the Lake Van area, snowmelt and rain peak from April to June and rising river discharge not only increases the lake level by about half a meter, but also delivers most of the clastics to the lake during this time of the year. At the same time, the mass of the dissolved ions is delivered to the lake by the spring-time freshwater spreading over the underlying saltier lake water. While clastics and organo-detritus settle directly to the lake bottom, dissolved ions concentrate during summer due to ongoing evaporation. Supersaturation necessary to cause CaCO3-precipitation is surpassed in late summer and autumn when the surface layer starts to mix with the higher saline lake water below. Aragonite and calcite constitute about 35 weight percent of the sediment (Landmann 1996). Thus, laminated sediments are deposited as varves with darker laminae in winter and spring and lighter carbonate dominated bands in late summer and autumn (Kempe 1977).
The Lake Van varves allow precise dating of sediments back to 14,740 a BP (Kempe 1977; Landmann et al. 1996a; Lemcke 1996; Landmann and Kempe 2005). The characteristics of each varve were scanned at high resolution enabling varve counting and automated processing by image analysis. Annual accumulation amounted to about 0.5 mm a−1 on average. Late Glacial chronozones are identified by abrupt changes of the annual accumulation rate coinciding with changes of chemical composition of the sediment. Chronozones at Lake Van correspond closely to those found in Greenland Ice cores indicating climatic teleconnection between the North Atlantic and Anatolia (Landmann et al. 1996b; Landmann and Kempe 2005).
Changes in the aragonite to calcite ratio, the presence of protodolomite and magnesite in certain profile sections, the long-term record of the annual accumulation rate, the water and pollen content of the sediment, the concentration of clastics, organic carbon, and opal and the texture of the sediment provide the basis to reconstruct the lake level history (Fig. 2; Landmann et al. 1996b). Terrace sediments up to 80 m above the present lake level (Schweizer 1975; Valeton 1978) constitute the oldest part of the sediment record. At Güselsu a terrace outcrop at 25 m above the present lake level yielded a floating varve chronology of 606 years fixed by 14C-dating to the period 20.1–20.7 cal. ka BP (Kempe et al. 2002). This varved section with an average accumulation rate of 0.9 cm a−1 represents the glacial high-stand of the lake at the end of the Late Glacial Maximum (LGM). It is underlain by sandy gravel marking the transgression of the glacial lake and overlain by a slumped, 6-m-thick section representing the high-stand and beginning regression of the lake. There is gap of about 5 ka between the floating chronology and the oldest sediment sampled in cores from the lake bottom. Sediments of the lowest section of cores K6 and K10 are not varved, they contain high content of dolomite and protodolomite, ooids, and rounded pumice pieces, all interpreted to reflect a shallow water environment with a water depth of only 30–40 m (Landmann et al. 1996b). If taking the current total salt content of the lake (i.e., 1.3 × 1013 kg, Reimer 1995) into account then NaCl should start precipitating once the lake volume is reduced to a depth less than 80 m (Fig. 2). However, some salt will be removed also by brine convection into the pore spaces of older sediments allowing further lake level lowering without a salinity crisis. Thus, the lake level appears to have dropped by more than 460 m within a period of 5 ka at the end of the LGM. Such a substantial drop can only have been caused by a drastic climate change toward warmer and drier conditions and must have left traces in other nearby sedimentary records as well. For example, the level of Lake Lisan, precursor of the Dead Sea, dropped simultaneously by at least 260 m causing precipitation of a massive gypsum bank called the ‘white member’ of the upper Lisan Formation (e.g., Landmann et al. 2002; Katz and Starinsky, this issue).
Apart from the post-LGM drop in lake level, three shorter recession periods are revealed by increased amounts of Mg-carbonates and aragonite in the sediment column (Landmann et al. 1996b; Lemcke 1996; Wick et al. 2003) coming along with significant decreases in porosity during these periods (Fig. 2). Strong lake level decreases also seem to be accompanied by a higher numbers of turbidites (Landmann 1996). All deep basin cores retrieved from water depths >396 m show very similar porosity profiles. In comparison, porosity in core K1 (340 m) is considerably lower, especially during the lake level high stand from 4–7 ka BP, indicating significant differences in the depositional conditions. The remarkably high variability of porosity in the younger sediments (<4 ka BP) is connected to alternating light and dark brown bands with lower and higher content of organic matter, respectively, that may reflect changing mixing behavior of the lake since that time.
The importance of Lake Van as a site for paleoclimate studies is now widely accepted. In May of 2006, a workshop was held by the International Continental Scientific Drilling Program to plan deeper drilling and programs for further research (Demirel-Schlüter et al. 2005).
4 Hydrochemistry of The Lake Water
Lake Van is a saline lake exhibiting a distinct soda chemistry defined by the fact that alkali cations, in particular sodium and potassium, maintain the charge balance of bicarbonate and carbonate ions in addition to alkaline earth ions (e.g., Kempe 1977; Kempe and Kaźmierczak 2003). Soda lakes show large salinity and pH ranges, but especially in closed lakes with high salt contents and alkalinities above 10−2 mol l−1 pH may rise to values above 10 (Eugster and Hardie 1978; Lerman and Stumm 1989; Kempe et al. 1989).
4.1 General Composition and The Carbonate System
One main effort of the expeditions in 1989 and 1990 was a comprehensive characterization of the soda chemistry of Lake Van including a detailed investigation of its hypolimnion. So far, analytical data as compiled in Table 1 were restricted to near-coast surface samples and to the samples taken down to 380 m in 1974 (Kempe 1977) that covered only a few of the important hydrochemical parameters. In spite of a relative large spread in results, the previous analyses showed that in Lake Van carbonate species are about 65-fold enriched compared to seawater and are overwhelmingly balanced by sodium and potassium. Chloride concentration is somewhat higher than the sum of bicarbonate and carbonate, but the absolute chloride concentration is three times lower than in the ocean. Sulfate concentration is comparable to seawater values. Concentrations of magnesium and calcium are low with calcium being extremely depleted.
Our June 1989 and 1990 profiles document the summer stratification caused by warming and (for the first time) by the annual supply of spring freshwaters as well as winter mixing of the epi- to mesolimnion (Fig. 3). High resolution CTD casts showed that both in winter 1988/89 and 1989/90 vertical mixing reached to a depth of about 70 m only (Wüest et al. 1993). T-S profiles revealed a warm (17–20°C) and less salty (21.9 g kg−1), about 10–15-m-thick surface layer. The layer below showed higher salinities and decreasing temperatures down to local minima of less than 3°C at 40 to 50 m depth, representing relicts of the winter cooling. From 40 to 100 m salinity increased to the deep water values of 22.5 g kg−1. The lower limit of deep water convection at 70–80 m was revealed by the local temperature maxima of 3.3–3.4°C and by the shape of the salinity gradients. In spite of the low salinity gradients in the hypolimnion, stratification was stable enough to compensate for the determined geothermal heat flux of about 0.10 W m−2 and exchange was dominated by turbulent diffusion with typical rates for a large lake in the order of 0.1–1.0 cm2 s−1 (Wüest et al. 1993; Kipfer et al. 1994).

Depth profiles of salinity, total alkalinity (TA), and major solutes in Lake Van in June 1989 and June 1990. Salinity: Conductivity-derived profils from Wüest et al. (1993) adjusted to calculated TDS and measured density, 1989: solid line, 1990: dashed line; TA and major ions 1989: black circles and triangles, 1990: open circles
Water column profiles of main ions illustrate the increase of salinity with depth (Fig. 3, Table 1). Lowest concentrations for total alkalinity, chloride, sulfate, sodium, potassium, and magnesium were found in the upper ten meters of the epilimnion, while calcium concentrations (0.105 mmol l−1) peaked in the epilimnion due to the influence of spring freshwater input. With the exception of calcium, concentrations increased downward toward the halocline. In 1990, compared to 1989, concentrations were higher at 20 to 70 m depth as a response to a lower springtime river input. In both years, major ion concentrations increased below the halocline along a hardly detectable gradient to deep water values, while calcium decreased to less than 0.09 mmol l−1 (Reimer 1995).
Because spring to early summer freshwater influx lowers total alkalinity in the surface layer, Lake Van displays an exceptional pH profile with depth (Fig. 4). Commonly, biological productivity during spring and summer would lead to an increase of pH by CO2 consumption in the surface layer, while pH would decrease in the hypolimnion due to CO2 release by remineralization of organic matter. Instead, even in summer the warm surface layer of Lake Van was characterized by lower pH values (9.73–9.74) compared to bottom water where pH increased to 9.87–9.88. From pH, temperature, and the appropriate ion concentrations saturation with respect to carbonate minerals, partial pressure of CO2 (PCO2), and other quantities of the carbonate system were computed with PHREEQE (Parkhurst et al. 1990; Schulz and Kölling 1992). Boron concentrations of 8.3–8.6 mmol l−1 were estimated from a fit of three single measurements (5, 50 and 400 m depth, Table 1) against salinity. Based on hydrometer measurements (20°C) on the samples from the depths above (Table 1) a density of 1.017 g cm−3 was applied for all sample depths in the PHREEQE calculations. Resulting mineral saturations are given as saturation index SImineral = log (IAP/Kmineral) (IAP = ion activity product; Kmineral = solubility product).

Depth profiles of temperature, pH, dissolved inorganic carbon (DIC), partial pressure of CO2 (PCO2), and saturation of calcite and dolomite calculated with PHREEQE in Lake Van in June 1989 (black circles and triangles) and June 1990 (open circles). Saturation is given as SI = log (ion activity product IAP/solubility product Kmineral)
The establishment of the warm surface layer during summer appears to be the single most important factor governing the distribution of the ionic species of the carbonate system with depth. Dissolved inorganic carbon (DIC) increased from 91 to 101 mmol kg−1 with depth and CO2(aq) concentrations (not shown) were calculated to be as low as 13 μmol kg−1 at the surface and 18 μmol kg−1 in bottom waters. Partial pressure of CO2 ranged from 260 μatm in the hypolimnion to values of 305–310 μatm in the surface layer that closely matched the atmospheric CO2 level at the altitude of the lake. Highest supersaturations with respect to calcite (SICalcite = 1.14), aragonite (SIAragonite = 0.99, not shown), and dolomite (SIDolomite = 3.85–4.10) were found in the upper 30–40 m of Lake Van and hypolimnion waters still exceeded calcite supersaturation 10-fold (Fig. 4). The shape of the dolomite saturation profile with depth coincided with the temperature profile and mirrored those of the carbonate system parameters as for example DIC distribution. Extreme high dolomite supersaturation values, i.e., exceeding the 104-fold of dolomite saturation, point to the common problem of kinetic inhibition of dolomite precipitation at low temperatures (Land 1998). Indeed, dolomite-like mineral phases were only exceptionally encountered in the sedimentary record of Lake Van, commonly occurring within deeper horizons related to evaporation events.
The curvature of calcite supersaturation mainly followed that of calcium concentration profiles reflecting that calcite saturation is controlled by the extremely low Ca2+ ion activities due to strong ion pairing rather then by the much larger carbonate pool. In spite of apparent high supersaturation, no carbonate precipitation in the central lake basin has been observed during sampling in June to July but ongoing evaporation during summer and early autumn is suggested to further enhance supersaturation of surface waters and initiate calcium carbonate precipitation. Basin-wide whitings as documented on satellite images from late summer to autumn (Landmann et al. 1993) may account for the annual deposition of calcium carbonate seen in the sedimentary record. In contrast, the effect of photosynthetic CO2 removal appears to be negligible in the well-buffered carbonate system in Lake Van (Arp et al. 2001).
4.2 Productivity, Nutrient Cycles, and Deep Water Renewal
Little is known about the biological productivity in Lake Van. Chlorophyll values of 0.1–0.9 μg l−1 in summer and autumn were reported by Tuğrul et al. (1984) and the normally high water transparency suggests moderate primary production rates. In both years, production in the epilimnion caused an oxygen maximum layer at depths between 10 and 50 m (Fig. 5), while concentrations decreased below the pycnocline reaching 17 μmol l−1 close to the water-sediment interface. Dissolved phosphate and dissolved silica were depleted in the surface layer (Fig. 5) in comparison to the concentrations of maximal 5.5 μmol l−1 phosphate and 130 μmol l−1 Si in deep water, but both were far from being completely consumed by photosynthesis. Especially, dissolved phosphate concentration of 3.5 μmol l−1 was still very high in the surface layer clearly pointing to nitrogen species as the production limiting nutrients in Lake Van. Unfortunately, the high alkaline lake water caused severe methodological difficulties regarding the reduction step in nitrate analysis. Measurements performed with a standard addition technique suggested that nitrate concentrations were lower than 1 μmol l−1 throughout the water column, while Tuğrul et al. (1984) reported concentrations of up to 10 μmol l−1 in the hypolimnion. With the exception of pronounced peaks in the lower epilimnion and higher concentrations in the bottom sample in 1989, where the Niskin Sampler most probably touched the sediment, nitrite and ammonia were close to or below detection limits of 0.02 μmol l−1 and 0.1 μmol l−1, respectively. Nitrite peaks of up to 0.7 μmol l−1 were recorded directly at the base of the oxygen maximum while an ammonia maximum of 0.4 μmol l−1 appeared some meters above the nitrite peak. Peaks in nitrite concentration at the base of the euphotic zone are commonly observed features in various stratified ocean basins and termed as the primary nitrite maximum which is either attributed to bacterial nitrification or nitrite release by phytoplankton. At the pycnocline the sinking velocity of particulate matter is reduced, and therefore enhanced microbial remineralization of particulate organic nitrogen or a zone of intense zooplankton grazing are discussed as possible explanations for the peak in ammonia concentration (for a recent review see Lomas and Lipschultz 2006).

Depth profiles of oxygen and nutrients in Lake Van in June 1989 (black circles and triangles) and June 1990 (open circles). Open squares: Oxygen distribution in September 1983 from Tuğrul et al. (1984)
In comparison to the oxygen depth distribution in 1989, an O2 deficiency of up to 50 μmol l−1 was observed in the depth range from 70 to 250 m in 1990, while corresponding phosphate concentrations were higher by about 0.2 μmol l−1 in 1990. Because vertical convection of the water column in winter 1989/1990 was restricted to the upper 70 m (Wüest et al. 1993), oxygen was not replenished in deeper waters and differences in concentration can be attributed to the aerobic remineralization of organic matter during a one year period. From integration over the depth range below the pycnocline total oxygen consumption amounted to about 2,700 mmol m−2 a−1, whereas about 14 mmol m−2 a−1 phosphate was released to the water column. The resulting molar O:P ratio of 193:1 was close to known biological production/remineralization ratios given by the Redfield–Ketchum–Richards ratio of O:C:N:P = 138:106:16:1 (Redfield et al. 1963) or newer investigations in the marine environment by Takahashi et al. (1985) and Anderson and Sarmiento (1994; O:C:N:P = 170:117:16:1). Equivalent increase in carbon concentration of deep water would have been not higher than 20 μmol kg−1 and therefore impossible to detect by chemical analysis at a DIC background concentration of 100 mmol kg−1 in the hypolimnion of Lake Van. Phosphate and silica curves correlate highly with each other, resulting in a P:Si mole ratio of 1:55.
From the above ratios and O2 consumption rates, annual carbon release was estimated to amount to 1,900–2,100 mmol m−2 a−1, or correspondingly to 23–25 g C m−2 a−1, which gives a first estimate of the carbon export production in Lake Van. Based on studies on primary production rates, export production, depth of the euphotic zone, and chlorophyll concentrations, Baines et al. (1994) suggested that export production comprises about 20–40% of the primary production in marine and limnic environments where primary production does not exceed 200 g C m−2 a−1 with maximum chlorophyll concentrations of 1 μg l−1. Empirical relations, connecting primary to export production were given by e.g., Eppley and Peterson (1979) or Berger et al. (1989). Introducing the derived carbon export production in these models yields a reasonable primary production rate of 100 g C m−2 a−1 for Lake Van that is in good agreement with the findings of Baines et al. (1994).
Gradients of oxygen and nutrients also delivered information on deep water renewal times in Lake Van. Kempe (1977) assumed lake convection to occur every year and Kipfer et al. (1994) concluded from He isotope and tritium diffusion/reaction models that deep waters of Lake Van are exchanged at least every second year. Contrary, CTD casts showed that both in winter 1988/89 and 1989/90 vertical mixing only reached to a depth of about 70 m (Wüest et al. 1993). Perennial stagnation of the hypolimnion of Lake Van is confirmed by the steep gradients of oxygen and phosphate. Tuğrul et al. (1984) observed oxygen distributions in autumn 1983 and summer 1984 that exhibited much higher concentrations of up to 240 μmol l−1 in the depth range from 50 to 250 m. If these high O2 concentrations are attributed to deep vertical mixing prior to 1983 the observed oxygen consumption until 1990, supported by the measured increase of phosphate in the upper hypolimnion from 1989 to 1990, most likely reflects ongoing remineralization of organic matter in deep water under continuous stagnant conditions for at least seven years. From turbulent diffusion rates Wüest et al. (1993) suggested that lack of deep water renewal in Lake Van for several years, while keeping the hypolimnion oxic, would require organic matter deposition rates not exceeding 15 g C m−2 a−1. In this case, the steep gradients of phosphate, and that of the comparatively similar dissolved silica in the deep hypolimnion below 300 m cannot be explained by remineralization of organic matter. Instead, additional fluxes from the lake bottom caused by the high pore water nutrient concentrations have to be considered.
5 Riverine Input
Expeditions in June/July 1989 and June/July 1990 included also a detailed study of lakes and springs in the vicinity of Lake Van and of its tributaries (Reimer 1995). In the Lake Van area, maximum precipitation occurs from March to April, but in case of a delayed snowmelt in the surrounding mountains rivers carry highest discharge in April and especially May. Runoff volume then is severalfold higher than the monthly mean discharge. Very low runoff is typical for the period August to the end of February. More than half of the annual runoff can be attributed to three rivers, the Zilan Cayi and Bendimahi Cayi discharging into the bay of Ercis and the Engil Cayi discharging into the bay of Van near the town of Gevas.
During sampling in late June 1989, riverine discharge was already low, indicative of the dry summer conditions in the Lake Van highlands. In contrast to this, the mountains around the lake were still covered with snow in early June 1990. River discharge was considerably higher than in the previous year and much more characteristic for the hydrologic situation commonly found in May during peak runoff.
Results of hydrochemical analysis are summarized in the Piper diagram of Fig. 6, revealing that bicarbonate is the dominant anion in the tributaries of Lake Van. Although most rivers belong to the Ca-Mg-bicarbonate type, in addition to alkaline earth elements frequently sodium and potassium also account for charge balance of the bicarbonate.

Hydrochemistry of rivers and sublacustrine springs discharging into Lake Van (1989 and 1990). 1 = Rivers of the Bitlis Mountains of the S/SE coast (Kücüksu, Gevas, Engil) and the region of Adilcevaz, 2 = Rivers of the NE/E coast (Karasu, Delicay, Süfresor), 3 = Zilan Cayi system, 4 = Bendimahi Cayi upstream and mixing zone (arrow denotes mixing trend), 5 = Sublacustrine springs near Resadiye and Tatvan Bay (arrow denotes mixing trend), 6 = Lake Van water (black diamonds)
River waters can be separated into classes reflecting the lithology of the respective drainage areas. The first group encloses bicarbonate/alkaline earth dominated waters, encompassing the rivers of the southern metamorphic Bitlis Mountains including the Engil Cayi at their southeastern foothills as well as the Murdasu River near Adilcevaz at the N-coast. Alkaline earth chemistry results from drainage of Cretaceous or Miocene limestones and ancillary dolomites, and erosion of Pleistocene carbonate terraces. In the estuary of the Murdasu River, that carries highest calcium concentrations of all investigated tributaries, extended whitings are a common feature. Spectacular whitings are also caused by the Kücüksu River in the Tatvan Bay to the southwest.
The second group encompasses rivers of the N and E coast which largely drain areas with volcanic rocks, but partly also limestone deposits that discharge predominantly bicarbonate waters. These rivers have in common that they carry higher amounts of alkali elements but total discharge, with the exception of the Karasu River and the Delicay, is low to intermediate.
The largest rivers, the Zilan Cayi with its tributaries and the Bendimahi Cayi, drain mostly volcanic areas and deliver about 40% of the annual total runoff of Lake Van into the bay of Ercis. The Zilan Cayi (forming group 3) has the lowest ion concentrations of all rivers but carries, because of its high annual discharge, together with the Bendimahi Cayi, about 50% of the annual dissolved chloride and sulfate load into Lake Van. The Bendimahi Cayi (group 4) alone is responsible for nearly 50% of the annual sodium and potassium load reflecting the peculiar river water chemistry where charge balance of bicarbonate is largely achieved by high alkali ion concentrations.
Because of the fact that runoff conditions and therefore also river hydrochemistry were different between the two years of the sampling, we used our analyses to estimate the total elemental annual inputs into Lake Van. Higher concentrations at low runoff in 1989 were used to estimate dissolved transport for the dry months from July to March, while high runoff and low concentrations in June 1990 were taken to represent conditions typically appearing from April to June. During sampling, discharge was determined at each sampling point at appropriate cross sections with a current meter. In addition, monthly mean discharge values from 1967 to 1985 available for gauge stations from all larger rivers were considered to calculate annual runoff. For rivers without gauge stations, annual variability was interpolated from direct flow measurements at the time of sampling and nearby stations. The procedure, further explained in Reimer (1995), summed up to an annual surface runoff of 1.81 km3, but did not include frequently observed influx of sublacustrine springs. Water balance calculation based on precipitation and evaporation rates and on the lake level curve suggests a slightly higher mean annual fresh water input in the order of 2 km3. Therefore, inputs of dissolved salts as calculated were increased by 10% to account for this discharge rate.
Comparison of the annual dissolved input with its total amount in Lake Van provided an estimate of accumulation times or “salt age” for each component (Table 2). Also, transport rates per lake area were set in relation to sedimentary deposition rates of various elements. Temporal changes in climate and hence runoff conditions and especially, possible loss of salts during evaporative cycles in lake history, could not be estimated. Therefore, this mass balance approach gives only a rough idea of the age of the lake system. Nevertheless, it points out some characteristics of element cycles in the lake.
All accumulation times are significantly higher than those calculated by Kempe (1977) based on the results of the 1974 sampling mostly because the recent total discharge estimate is 25% lower than previously used. Additionally, the 1974 sample for the Bendimahi Cayi, the river with the highest discharge, apparently was taken near its mouth and was probably influenced by Lake Van water. Therefore, input rates for this river were overestimated especially for chloride, sodium, and potassium. Chloride has the highest salt age of about 200 ka (Table 2); a result close to the age discussed for the closure of the Mus valley by the Nemrut ignimbrite as a precursor for Lake Van formation (Özdemir et al. 2006) and also to the proposed onset of tectonic pull-down of the basin given by Wong et al. (1978) and Degens et al. (1984). Accumulation times are significantly lower for sodium and boron ranging in the order of 100 ka. Next is sulfur (60 ka) and potassium (30 ka) while all other elements yielded ages <10 ka. Calcium has, with 82,000 t a−1, the highest input rate, corresponding to 23 g m−2 a−1, but it also has the lowest salt age with 27 years, because it is quantitatively precipitated as carbonate. The calculated real deposition rate based on the analyses of our sediment cores over the last 4 ka yielded a flux of 19.8 g m2 a−1 that is very similar to the mass balance value. Dilute inflows into Lake Van are characterized by an average molar Mg to Ca ratio of 2:1. Due to the persistent loss of calcium the molar Mg:Ca ratio in the lake water increased to recent values of between 40:1 and 50:1 in the surface water and the bottom water, respectively. Magnesium is also depleted in the lake water, yielding a salt age of 1.87 ka. Since the amount of Mg-carbonates in the sediment (Landmann 1996; Lemcke 1996) is small, Mg is most probably removed with amorphous Mg-silica phases as suggested from pore water and sediment geochemistry (see below, and Reimer 1995). While depletion of potassium relative to chloride could be explained by ion exchange or formation of authigenic clay minerals in the sediment other mechanisms must be responsible for the removal of sodium, sulfur, and carbon. Neither sulfate reduction nor precipitation of gypsum (not possible because of the extremely low Ca2+ concentration) can currently account for the relative depletion of sulfate. Virtually no pyrite is found in sediments of Lake Van, the formation of which may be prevented by supersaturation of Fe-monosulfides under highly alkaline conditions as suggested by Domagalski et al. (1990) for Mono Lake and Walker Lake. Also, accumulation time of dissolved inorganic carbon is very low. Nevertheless, deposition of Ca-carbonate and organic carbon in the sediments can only account for half of the annual influx by rivers. Thus, there must have been different mechanisms working in the past too.
Accumulation times for nutrients are generally very low indicating fast biological fixation and loss to the sediment. Remarkably, sedimentary deposition rates of P, N, and Si, being rather constant throughout the last 4 ka, are higher than the recent annual river influx of dissolved constituents. As supported especially by the steep concentration gradients of phosphate and dissolved silica in the hypolimnion of Lake Van, replenishment by fluxes out of the sediment rather than recent river transport balances nutrient concentration levels within the lake.
6 Chemistry of Pore Water
Pore water chemistry was analyzed in detail on cores taken from the central Tatvan basin (K2, 435 m depth), the southern slope-basin transition (K6, 396 m depth), and the western slope of the Tatvan fan (K1, 340 m depth). Upon opening, grayish-black sediment color, strong H2S smell, and Eh values decreasing from −150 mV in the top layers to −250 mV in deeper parts of the core showed that the zone of sulfate reduction starts immediately below the water-sediment interface. The pH values decrease slightly below the sediment surface, presumably due to release of carbon dioxide by ongoing decomposition of organic matter or to reverse weathering reactions like neoformation of Mg-silica-rich mineral phases as discussed below.
In all cores, pore water salinity increases with sediment depth reflecting upward diffusion of salts. However, concentration gradients of main ions are different between cores and obviously depend on the water depth of core recovery (Fig. 7). Within the overall linear increase of major ion concentrations downcore there are some minor excursions of the gradients at greater depths where porosity is noticeably lower in sediment sections older than about 10.5 ka BP. Also, gradients are steeper in the near surface section of K6, most probably resulting from a slump sequence inclosing higher pore water concentrations that replaced part of the ordinary sedimentary succession.

Distribution of pH, total alkalinity (TA), and major elements in pore water of cores from different water depths. K1: 340 m (open circles), K6: 396 m (black squares), K2: 435 m (black triangles). Open triangles denote lake bottom water concentrations. pH was measured at laboratory temperature of 20 ± 3°C. The shaded area marks a sediment section of core K6 that was replaced by older slump sediments. The differences in concentration especially in the upper core sections between core K6 and K2 are attributed to this slump (see text)
In the deepest pore water samples of cores K2 and K6 from 396 m and 435 m water depth, Cl-, Na-, and K-concentrations reach values about twice as high as in the lake. Total alkalinity increases 2.8-fold in core K2 and maximum density, obtained from a simple gravimetrical determination, is about 1.05 g cm−3. In the upper section of core K2, sulfate reduction accounts for a minor depletion of sulfate that is counterbalanced by sulfate diffusion from the downcore section. This strong increase of concentrations in the deep basin cores is attributed to a past desiccation phase with formation of concentrated brines now diffusing to the surface, thereby resembling the situation reported by Domagalski et al. (1990) for the central basin of Walker Lake in California. In contrast, salt gradients in core K1 from 340 m water depth are much lower. Total alkalinity as well as Cl-, Na-, and K-concentrations increase only up to 1.3-fold downcore. Sulfate depletion by sulfate reduction is more pronounced in this core since diffusive fluxes from below are less important.
To a certain extent, remineralization of deposited organic matter may explain the increase of total alkalinity (coming along with the increase of DIC). This view is substantiated by the distinct simultaneous release of phosphate and ammonia (Fig. 8) that, in case of core K1 from the shallower water depth, is characterized by rather constant molar C:N and C:P ratios of 18 and 165, respectively. However, alkalinity and phosphate concentration increase in excess to ammonia in the cores from the deep basin. Also, absolute concentrations are much higher in basin cores with total alkalinity reaching 440 mmol l−1 and phosphate 1 mmol l−1 at the bottom of core K2 which for phosphate is 170 times the recent concentration in bottom waters of Lake Van. Strong increases in alkalinity and phosphate during sulfate reduction are commonly reported (e.g., Krom and Sholkovitz 1977; Niewöhner et al. 1998; Domagalski et al. 1990, Kempe 1990), but the observed release is hardly balanced by the required input of organic matter as deduced from concentrations of organic carbon (2–5 mol kg−1) and organically bound phosphorus (10–15 mmol kg−1) in the upper sediment column. Decomposition of organic matter, therefore, may only in part account for the observed high concentrations and gradients in the cores from the deep basin, and the increase in alkalinity and phosphate is rather driven by both sulfate reduction and the diffusive delivery from deep, concentrated pore fluids.

Distribution of nutrients, calcium, magnesium, dissolved silica, and saturation of calcite and hydroxy-apatite calculated with PHREEQE in pore water of cores from different water depth. K1: 340 m (open circles), K6: 396 m (black squares), K2: 435 m (black triangles). Open triangles denote lake bottom water concentrations. Saturation is given as SI = log (ion activity product IAP/solubility product Kmineral). The shaded area marks the depth of the slump section in core K6
In contrast to concentrations of anions and alkali cations, which all show linear increases downcore, concentrations of the alkaline earth elements Mg and Ca decrease within the uppermost meter of cores K1, K2, and K6, indicating that these ions are removed from the pore water and fixed in the surficial sediments (Fig. 8). Ca concentrations drop to values of 0.05–0.06 mmol l−1 and accordingly calculated calcite saturations slightly decrease, eventually reflecting precipitation of CaCO3 in the uppermost sediment. On the other hand, the steady increase of phosphate concentrations downcore may cause the precipitation of a Ca-phosphate phase, thereby also reducing the dissolved Ca content of the pore water. For comparison, the saturation of hydroxy-apatite was calculated (Fig. 8). It is saturated in the lower parts of the deeper cores, but X-ray diffraction data did not record any apatite in the sediment, possibly due to the expected very low concentration.
The short accumulation time of magnesium (Table 2) already indicated that there is an effective removal mechanism for Mg at work in Lake Van sediments. Pore water profiles display a distinct decrease in dissolved Mg within the uppermost 2 m of all cores (Fig. 8). Even lower values are reached within a 5-m-long horizon in the two deep basin cores at depths representing sediment ages from about 11–2.5 ka BP. Mg fluxes at the water-sediment interface were estimated applying a temperature and tortuosity corrected diffusion coefficient Dsed-Mg of 2.5 × 10−6 cm2 s−1 and a porosity of 90% (Boudreau 1996). The calculated fluxes of 3–9 g Mg m−2 a−1 suggest a Mg fixation rate close to the estimated annual river input, thus explaining the short accumulation time of Mg in Lake Van.
X-ray diffraction data reveal the formation of Mg-carbonates only during periods of lake regression thus excluding this process as permanent sink for magnesium (Landmann et al. 1996b; Lemcke 1996). According to Khoo et al. (1978) sediments in Lake Van to a large amount consist of authigenic mixed-layer clay minerals. Trioktaedric smectites are suggested by Jones and Weir (1983) and Jones (1986) to be the stabile phase under alkaline conditions. Most probably, decrease of Mg concentrations may either be caused by Mg-adsorption on clay mineral surfaces or neoformation of authigenic mineral phases where contemporaneous dissolution of siliceous microorganisms such as diatoms is commonly regarded as a silica source (Badaut and Risacher 1983; Risacher and Fritz 1991).
The distribution of dissolved silica with sediment depth is much different from those of the other dissolved compounds (Fig. 8). Cores of the deep basin show broad plateaus of maximum concentrations in the mid sediment section where in the deepest core, K2, dissolved silica concentrations are stable at 2 mmol l−1 over a core distance of about 2 m. The maximum is less pronounced and not as long-lasting in core K6 from 396 m depth that embeds a slumped sediment section. The maximum in dissolved silica concentrations does not occur at all in the shallower core K1 from 340 m depth where concentrations stay nearly constant at 0.2 mmol l−1. In correspondence to this, the extractable opaline silica, for comparison of the exact stratigraphic position in the different cores plotted against sediment age in Fig. 9, shows also highest concentrations at the times when the dissolved silica concentration peaks. Opaline silica normally constitutes 2 mol kg−1 on average of the sediment or 15 wt% if calculated as SiO2 × 0.4 H2O with 10% bound water according to Mortlock and Froelich (1979). But it exceeds 5 mol kg−1 (40 wt% SiO2 × 0.4 H2O) in the maxima of the deep basin cores. The peaks in opaline silica are more pronounced than the dissolved silica maxima that are obviously broadened by diffusion in the pore water. Furthermore, a minimum opaline silica content occurred at 10.7 ka BP in K6 corresponding to the regression in lake level at that time.

Distribution with age of extractable opaline silica, dissolved silica, and saturation of amorphous silica calculated with PHREEQE in pore water of cores from different water depth. K1: 340 m (open circles), K6: 396 m (black squares), K2: 435 m (black triangles). Saturation is given as SI = log (ion activity product IAP/solubility product Kmineral). The shaded area marks the depth of the slump section in core K6, accentuating the cut-off of the maximum in opaline silica in this core
Legler and Krasske (1940) and Gessner (1957) described numerous diatom species from Lake Van but, when investigating the 1974 cores, Golubic and Buch (1978) found diatoms only in sediment from very shallow water or in samples from a certain sequence of core 13 taken from 400 m water depth in the Tatvan Basin (Fig. 1). Golubic and Buch (1978) argued that diatoms in the highly alkaline environment were only preserved in the sediment if buried fast within turbidites or below slumps. This conclusion, however, cannot be maintained since diatom frustules would also have been dissolved in alkaline pore waters. Instead, the sediment sequence in which diatoms are preserved clearly coincides with maximum opaline silica concentrations, spanning from 6.2–3.7 ka BP, in the sediments of the deep Tatvan basin (Fig. 9). Here, X-ray diffraction analyses demonstrated the presence of broad reflections characteristic for opal-A modifications, but in the remaining sediment column distinct mineral phases as a source of the extractable, still high background concentrations of opaline silica could not be identified (Landmann 1996).
Calculation of the saturation index of amorphous silica shows that for much of the plateau in dissolved silica in core K2 and for the maximum in K6 saturation is reached (Fig. 9). Ongoing dissolution of frustules and diffusive upward and downward transport of dissolved silica have led to broadening of the dissolved silica maxima, which is much smaller in core K6 where the upper part of the diatom sequence is replaced by slump sediments (Fig. 9). Propagation of the diffusive silica gradients clearly coincides with the extended minima of the Mg pore water concentration in cores K2 and K6 between depths corresponding to sediment ages of 11 and 2.5 ka BP. The coherence between the shape of the dissolved silica and Mg distribution in fact suggests the formation of an authigenic magnesium-silica phase in this sediment sequence of both basin cores, in addition to the Mg-fixation that already happens close to the sediment water interface.
The ratio of magnesium to silica fixation was estimated from calculation of fluxes at the upper and lower boundaries of the Mg minimum and dissolved silica maximum applying corrected diffusion coefficients of Dsed-Mg 2.5 × 10−6 cm2 s−1, Dsed-Si 3.3 × 10−6 cm2 s−1, and porosities of 90% and 80%, respectively. The resulting molar Mg:Si ratios amount to 0.69 for core K6 and to 0.59 in case of K2. These values differ only slightly from ratios reported for the experimental precipitation of Mg-hydroxysilicates under high pH conditions (Mg:Si = 0.75) by Kent and Kastner (1985) and the ideal ratio for sepiolite (Mg:Si = 0.67). Hardie and Eugster (1970) and Eugster and Hardie (1978) suggested sepiolite as an early diagenetic phase rich in Mg and Si, which on the other hand could not be identified in many recent saline lakes (e.g., Jones 1984; Spencer et al. 1985). Instead, Spencer et al. described an authigenic phase with a molar Mg:Si ratio of 1:1 to occur in the upper sediment of Great Salt Lake. Similar phases have recently been reported from quasi-marine, alkaline Lake Satonda, Indonesia, where they occur within microbialitic carbonates (Arp et al. 2003). From the above, it is supposed that such rather labile early diagenetic Mg–Si-phases are the source of the high background concentrations of extractable opaline silica in Lake Van sediments and therefore their neoformation is a major control on magnesium cycling in Lake Van.
7 Conclusions
The sedimentary record and the study of the pore water chemistry indicate that the geochemical evolution of Lake Van is governed by major rapid desiccation and transgressional phases throughout the Holocene and late Pleistocene.
In order to reconstruct the post-glacial history of Lake Van and its water body, we must explain several striking facts resulting from the investigations of the sediment, pore water, and lake water chemistry. These facts are:
-
i.
The downcore linear increase in chloride, sodium, potassium, and total alkalinity in the cores of the deep basin;
-
ii.
the missing of this increases in core K1 from 340 depth;
-
iii.
the congruence of dissolved silica with layers of preserved diatoms in the deep cores;
-
iv.
the interdependence of the magnesium and silica chemistry; and
-
v.
the large difference in the residual ages of the dissolved constituents in Lake Van.
Sediment and pore water chemistry, in particular the dissimilarities between the cores and the high salinities in the cores from the basin, can best be explained by assuming that the water column of Lake Van became stratified subsequent to the last prominent regression culminating at around 10.7 ka BP when lake level had been reduced to ca. 140 m and, in turn, salinity might have increased 8-fold. We suggest that during the following Holocene transgression the lake only partially mixed and kept a higher saline water body in the deep basin that existed for about 7 ka. The pycnocline of this deep basin brine (DBB) should have been placed at a water depth somewhere between 340 m (K1) and 396 m (K6) to account for the different evolution of the diffusion gradients in these cores from the slope and the basin with higher concentrations preserved in the deep sediments overlain by the DBB.
The existence of a permanently anoxic DBB is also indicated by very low amounts of deposited particulate Mn and correspondingly high Fe:Mn ratios for the time period of 10−3.2 ka BP as reported by Lemcke (1996), since under anaerobic conditions Mn becomes mobile and the tendency of Mn deposition decreases (Stumm and Morgan 1996). Starting with 3.2 ka BP total Mn considerably increased depicting a subsequent flip-flop like pattern that is attributed to vanishing of the DBB and consecutively varying redox condition at the sediment-water interface.
An anoxic DBB would further explain the preservation of the diatomaceous ooze in basin cores (K2, K6) while it is completely missing in the core from the slope (K1). There is no reason to assume that sedimentation rates of diatoms varied significantly at the different positions. Diatoms that reached the lake bottom within the overlying less saline water body should rapidly have been dissolved at the sediment-water interface as comparable to recent conditions. The diatoms reaching the DBB would also be dissolved, but the missing mixing would lead to a steady increase in dissolved silica. When the saturation limit of amorphous silica was reached in the DBB, settling diatoms started to be preserved in the sediments. This happened shortly after 7 ka BP. The concentration of extractable opaline silica returned to background values at around 3.7 ka BP, advertising the loss of the preservation potential of diatoms caused by the final overturn of the lake and the vanishing of the DBB. This event was most probably triggered by the arid period beginning at around 4−3.5 ka BP during which the lake level dropped from its highest stand during the Holocene climatic optimum by about 40 m to its recent level (Landmann et al. 1996a, b; Lemcke 1996; Wick et al. 2003). Since that time, the deep water body of Lake Van seems to switch between oxic and anoxic conditions with a frequency of 10 to 100 years, suggested by the pronounced color banding in the late Holocene sedimentary record.
Investigation of the lake’s hydrochemistry and its tributary rivers allowed calculating accumulation times for dissolved elements. The shorter these times are, the faster the constituents become removed by geochemical processes. In general, the compositional history of the lake water follows the evolutionary path toward Na–CO3–Cl–(SO4) brines as outlined by Garrels and Mackenzie (1967) and by Hardie and Eugster (1970), Eugster and Hardie (1978), and Eugster and Jones (1979) for alkaline lakes of the East African rift valley and the western United States, and most recently by Risacher and Fritz (this issue) for alkaline Bolivian salars.
Dilute inflows into Lake Van are characterized by predominantly bicarbonate waters with alkaline cations slightly exceeding alkaline earth cations. Incoming Ca is almost completely lost by precipitation of calcite and aragonite, and in the past has been lost also as protodolomite during desiccation events. Mg-depletion is caused by the neoformation of clay minerals or Mg-silica-rich precursors in the sediments. Mg-fixation largely occurs in the top layer of sediment and appears to be controlled by delivery and dissolution rates of diatoms emphasizing that magnesium and silica cycles in Lake Van are closely related.
Chloride is the most conservative element in Lake Van. In comparison to it fractionation is also obvious for other solutes such as K, Na, SO4, and DIC. Relative loss of these solutes is largest for K but a process that withdraws K from the lake water was not identified. Instead, pore water profiles actually depict diffusive replenishment into the lake, rather than reflecting K-adsorption to or K-exchange with clay minerals.
The depletion of sulfate also poses a problem. Although SO4 reduction leads to an increase of H2S and formation of metastable Fe-monosulfides as observed from the black colored sediments their amount would hardly balance the SO4 deficit. Similarly, carbonate deposition rates show that this process can only account for part of the calculated loss of DIC. Thus, there must have been other mechanisms that removed sulfur and carbon from the lake compared to the conservative Cl. We suggest that additional losses of dissolved sulfur and carbon could have occurred during the overturn of the lake. The anaerobic DBB would also have accumulated H2S from sulfate reduction and a certain amount of excess CO2 and methane from the disintegration of organic matter. These gases could have been released from the lake during the final overturn, thus helping to explain the SO4 and DIC deficits.
Another possibility is that Na- or even K-carbonate or sulfate salts precipitated during periods of drastically enhanced evaporation in lake history. Based on the recent lake chemistry, supersaturation with respect to minerals such as Na-carbonates or burkeite (Eugster and Hardie 1978) would demand evaporation down to 5% of the recent lake volume. This would correspond to a lake level drawdown by 350 m that has indeed been supposed from the sedimentary record for the late Pleistocene (Landmann et al. 1996b).
Overall the investigations presented here show that there are still many open questions as to history of Lake Van since its Last Glacial high stand. We are far from understanding even Lake Van’s major element balances. This is not only of importance for the lake itself, but for our understanding of the development of alkaline lake systems in general with implications to hypothesize about the chemical development of the primordial ocean and life itself.
References
Abich H (1856) Vergleichende chemische Untersuchung der Wässer des Caspischen Meeres, Urmia- und Van See. MAISSP, série 6, t.7:1–57
Anderson LA, Sarmiento JL (1994) Redfield ratios of remineralization determined by nutrient data analysis. Global Biogeochem Cycles 8:65–80. doi:10.1029/93GB03318
Arp G, Reimer A, Reitner J (2001) Photosynthesis-induced biofilm calcification and calcium concentrations in Phanerozoic oceans. Science 292:1701–1704. doi:10.1126/science.1057204
Arp G, Reimer A, Reitner J (2003) Microbial reef formation in seawater of increased alkalinity, Satonda Crater Lake, Indonesia. J Sed Res 73:105–127. doi:10.1306/071002730105
Badaut D, Risacher F (1983) Authigenic smectite on diatom frustules in Bolivian saline lakes. Geochim Cosmochim Acta 47:363–375. doi:10.1016/0016-7037(83)90259-4
Baines SB, Pace ML, Karl DM (1994) Why does the relationship between sinking flux and planktonic primary production differ between lakes and oceans? Limnol Oceanogr 39:213–226
Berger WH, Smetacek VS, Wefer G (1989) Ocean productivity and paleoproductivity—an overview. In: Berger WH, Smetacek VS, Wefer G (eds) Productivity of the ocean: past and present. Wiley, New York, pp 1–34
Boudreau BP (1996) Diagenetic models and their implementation. Springer, Berlin, New York, 414 pp
Danulat E, Kempe S (1992) Nitrogenous waste excretion at extremely alkaline pH: the story of Chalcalburnus tarichi (Cyprinidae), endemic to Lake Van, Eastern Turkey. Fish Physiol Biochem 9:377–386. doi:10.1007/BF02274218
Degens ET, Kurtman F (eds) (1978) Geology of Lake Van. Miner Res Explor Inst Turkey 169, Ankara, 158 pp
Degens ET, Wong HK, Kempe S, Kurtmann F (1984) A geological study of Lake Van, eastern Turkey. Geol Rundsch 73:701–773. doi:10.1007/BF01824978
Demirel-Schlüter F, Krastel S, Niessen F, Demirbag E, Imren C, Toker M, Litt T, Sturm M (2005) Seismic pre-site survey for a potential new ICDP site—PaleoVan—at Lake Van, Turkey. Geophys Res Abstr 7:J-07997
Directorate for Hydrography and Oceanography (1985) Van Gölü Sea Chart
Domagalski JL, Eugster HP, Jones BF (1990) Trace metal geochemistry of Walker, Mono and Great Salt Lakes. In: Spencer RJ, Chou I-M (eds) Fluid-mineral interactions: a tribute to HP Eugster, vol 2. Geochem Soc Spec Publ, pp 315–353
Eppley RW, Peterson B (1979) Particulate organic matter flux and planktonic new production in the deep ocean. Nature 282:677–680. doi:10.1038/282677a0
Eugster HP, Hardie LA (1978) Saline lakes. In: Lerman A (ed) Lakes: chemistry geology physics. Springer, Berlin, pp 237–293
Eugster HP, Jones BF (1979) Behavior of major solutes during closed-basin brine evolution. Am J Sci 279:609–631
Garrels RM, Mackenzie FT (1967) Origin of the chemical composition of some springs and lakes. In: Equilibrium concepts of natural water systems. American Chemical Society, Advances in Chemistry 67:222–242
Gessner F (1957) Van Gölü. Zur Limnologie des großen Soda-Sees in Ostanatolien (Türkei). Arch Hydrobiol 53:1–22
Golubic S, Buch B (1978) Diatoms in Lake Van Sediments. In: Degens ET, Kurtman F (eds) Geology of Lake Van. Miner Res Explor Inst Turkey 169, Ankara, pp 111–114
Grasshoff K, Ehrhardt M, Kremling K (1983) Methods of seawater analysis. Verlag Chemie, Weinheim, pp 125–187
Hardie LA, Eugster HP (1970) The evolution of closed-basin brines. Miner Soc Am Spec Pap 3:273–290
Irion G (1973) Die anatolischen Salzseen, ihr Chemismus und die Entstehung ihrer chemischen Sedimente. Arch Hydrobiol 71:517–557
Jones BF (1984) Clay mineral diagenesis in lacustrial environments. In: Bodine MW (ed) Workshop on diagenesis. US Geol Surv Bull 1490:100–127
Jones BF (1986) Clay mineral diagenesis in lacustrine sediments. US Geol Surv Bull 1578:291–300
Jones BF, Weir AH (1983) Clay minerals of Lake Abert, an alkaline, saline lake. Clays Clay Miner 31:161–172. doi:10.1346/CCMN.1983.0310301
Katz A, Starinsky A Geochemical evolution of the Dead Sea. Aquat Geochem (this issue)
Kaźmierczak J, Kempe S (2003) Modern terrestrial analogues for the carbonate globules in Martian meteorite ALH84001. Naturwissenschaften 90:167–172
Kempe S (1977) Hydrographie, Warvenchronologie und organische Geochemie des Van Sees, Osttürkei. Dissertation, Mitt Geol-Paläont Inst Univ Hamburg 47:125–228
Kempe S (1990) Alkalinity: the link between anaerobic basins and shallow water carbonates? Naturwissenschaften 77:426–427. doi:10.1007/BF01135940
Kempe S, Degens ET (1985) An early soda ocean? Chem Geol 53:95–108. doi:10.1016/0009-2541(85)90023-3
Kempe S, Degens ET (1978) Lake Van varve record: the past 10,420 years. In: Degens ET, Kurtman F (eds) Geology of Lake Van. Miner Res Explor Inst Turkey 169, Ankara, pp 56–63
Kempe S, Kaźmierczak J (1994) The role of alkalinity in the evolution of ocean chemistry, organization of living systems and biocalcification processes. In: Doumenge F (ed) Past and present biomineralization processes. Considerations about the carbonate cycle, Bull Inst Oceanogr, Monaco, no. spec. 13:61–117
Kempe S, Kaźmierczak J (2002) Biogenesis and early life on Earth and Europa: favored by an alkaline ocean? Astrobiology 2(1):123–130. doi:10.1089/153110702753621394
Kempe S, Kaźmierczak J (2003) Modern soda lakes: model environments for an early alkaline ocean. In: Müller T, Müller H (eds) Modelling in natural sciences; design, validation and case studies. Springer, Berlin, pp 309–322
Kempe S, Kaźmierczak J (2007) Hydrochemical key to the genesis of calcareous non-laminated and laminated cyanobacterial microbialites. In: Seckbach J (ed) Extremophilic algae, cyanobacteria and non-photosynthetic protists: from procaryots to astrobiology. Springer, Berlin, COLE Series, pp 241–264
Kempe S, Kaźmierczak J, Degens ET (1989) The soda ocean concept and its bearing on biotic and crustal evolution. In: Crick RE (ed) Origin, evolution and modern aspects of biomineralization in plants and animals. Proceedings of 5th International Symposium on biomineralization, Arlington, Texas, May, 1986, Plenum Press, New York, pp 29–43
Kempe S, Kaźmierczak J, Landmann G, Konuk T, Reimer A, Lipp A (1991) Largest known microbialites discovered in Lake Van, Turkey. Nature 349:605–608. doi:10.1038/349605a0
Kempe S, Landmann G, Müller G (2002) A floating varve chronology from the Last Glacial Maximum terrace of Lake Van/Turkey. Z Geomorphol 126:97–114
Kent DB, Kastner M (1985) Mg2+ removal in the system Mg2+—amorphous SiO2–H2O by adsorption and Mg-hydroxysilicate precipitation. Geochim Cosmochim Acta 49:1123–1136. doi:10.1016/0016-7037(85)90003-1
Khoo F, Degens ET, Lambert A (1978) Geochemistry of Lake Van sediments. In: Degens ET, Kurtman F (eds) Geology of Lake Van. Miner Res Explor Inst Turkey 169, Ankara, pp 81–91
Kipfer R, Aeschbach-Hertig W, Baur H, Hofer M, Imboden DM, Signer P (1994) Injection of mantle type helium into Lake Van (Turkey): the clue for quantifying deep water renewal. Earth Planet Sci Lett 125:357–370. doi:10.1016/0012-821X(94)90226-7
Krom MD, Sholkovitz ER (1977) Nature and reactions of dissolved organic matter in the interstitial waters of marine sediments. Geochim Cosmochim Acta 41:1565–1573. doi:10.1016/0016-7037(77)90168-5
Land LS (1998) Failure to precipitate dolomite at 25°C from dilute solution despite 1000-fold oversaturation after 32 years. Aquat Geochem 4:361–368. doi:10.1023/A:1009688315854
Landmann G (1996) Van See/Türkei: Sedimentologie, Warvenchronologie und Paläoklima der letzten 15 000 Jahre. Dissertation, Facult Geosci Univ. Hamburg, 137 pp, unpublished
Landmann G, Kempe S (2005) Annual deposition signal versus lake dynamics: microprobe analysis of laminated Lake Van (Turkey) sediments reveals missing varves in the period 11.2–10.2 ka BP. Facies 51:135–145. doi:10.1007/s10347-005-0062-9
Landmann G, Kempe S, Reimer A (1993) Bathymetrie und Satellitenaufnahmen. Kap. 2 in Abschlußbericht, DFG Projekt Wo 395/2-1 bis 2-4. Inst Biogeochem Meereschem Univ Hamburg, 3 pp (unpublished)
Landmann G, Reimer A, Lemcke G, Kempe S (1996a) Dating Late Glacial abrupt climate changes in the 14, 570 years long continuous varve record of Lake Van/Turkey. Paleogeogr Paleocl Paleoecol 122:107–118. doi:10.1016/0031-0182(95)00101-8
Landmann G, Reimer A, Kempe S (1996b) Climatic induced lake level changes of Lake Van/Turkey during the transition Pleistocene/Holocene. Global Biogeochem Cycles 10(4):797–808. doi:10.1029/96GB02347
Landmann G, Abu Qudaira GM, Shawabkeh K, Wrede V, Kempe S (2002) Geochemistry of Lisan and Damya formation in Jordan and implications on palaeoclimate. Quaternary Int 89(1):45–57. doi:10.1016/S1040-6182(01)00080-5
Langbein WB (1961) Salinity and hydrology of closed lakes. US Geol Surv Pap 412:1–19
Legler F, Krasske G (1940) Diatomeen aus dem Van See (Armenien). Beiträge zur Ökologie der Brackwasserdiatomeen. Beih Bot Cbl 60:335
Lemcke G (1996) Paläoklimarekonstruktion am Van See (Ostanatolien, Türkei). Dissertation 11786, 182 pp, Eid Techn Hochsch, Selbstverlag
Lerman A, Stumm W (1989) CO2 storage and alkalinity trends in lakes. Water Res 23:139–146. doi:10.1016/0043-1354(89)90037-7
Liebezeit G (1991) Analytical phosphorus fractionation of sediment trap material. Mar Chem 33:61–69. doi:10.1016/0304-4203(91)90057-4
Livingstone DA (1963) Chemical composition of rivers and lakes. US Geol Surv Prof Pap 440-G:1–61
Lomas MW, Lipschultz F (2006) Forming the primary nitrite maximum: nitrifiers or phytoplankton? Limnol Oceanogr 51(5):2453–2467
López-García P, Kaźmierczak J, Benzerara K, Kempe S, Guyot F, Moreira D (2005) Bacterial diversity and carbonate precipitation in the microbialites of the highly alkaline Lake Van, Turkey. Extremophiles 9:263–274. doi:10.1007/s00792-005-0457-0
Millero FJ (1979) The thermodynamics of the carbonate system in seawater. Geochim Cosmochim Acta 43:1651–1661. doi:10.1016/0016-7037(79)90184-4
Mortlock RA, Froelich PN (1989) A simple method for the rapid determination of biogenic opal in pelagic marine sediments. Deep-Sea Res 26:1415–1426. doi:10.1016/0198-0149(89)90092-7
Müller G, Irion G, Förstner U (1972) Formation and diagenesis of inorganic Ca-Mg carbonates in the lacustrine environment. Naturwissenschaften 59:159–164
Niewöhner C, Hensen K, Kasten S, Zabel M, Schulz HD (1998) Deep sulfate reduction mediated by anaerobic methane oxidation in sediments of the upwelling area off Namibia. Geochim Cosmochim Acta 62:455–464. doi:10.1016/S0016-7037(98)00055-6
Özdemir Y, Karaoğlu Ö, Tolluoğlu AÜ, Güleç N (2006) Volcanostratigraphy and petrogenesis of the Nemrut stratovolcano (East Anatolian High Plateau): the most recent post-collisional volcanism in Turkey. Chem Geol 226:189–211. doi:10.1016/j.chemgeo.2005.09.020
Parkhurst DL, Thorstenson DC, Plummer LN (1990) PHREEQE—a computer program for geochemical calculations. (Conversion and upgrade of the prime version of PHREEQE to IBM PC-compatible systems by Tirisanni JV, Glynn PD). US Geol Surv Wat Res Invest Rep 80–96:1–197
Parrot J (1834) Reise zum Ararat. 2 Teile. Haude & Spener, Berlin, pp 262 and 198
Redfield AC, Ketchum BH, Richards FA (1963) The influence of organisms on the composition of sea water. In: Hill MN (ed) The sea, vol 3. Wiley, New York, pp 26–77
Reimer A (1995) Hydrochemie und Geochemie der Sedimente und Porenwässer des hochalkalinen Van Sees in der Osttürkei. Dissertation, Facult Geosci Univ Hamburg, 136 pp, unpublished
Risacher F, Fritz B (1991) Geochemistry of Bolivian salars, Lipez, southern Altiplano: origin of solutes and brine evolution. Geochim Cosmochim Acta 55:687–705. doi:10.1016/0016-7037(91)90334-2
Risacher F, Fritz B Origin of salts and brine evolution of Bolivian and Chilean Salars. Aquat Geochem (this issue)
Schulz HD, Kölling M (1992) Grundlagen und Anwendungsmöglichkeiten hydrogeochemischer Modellprogramme. DVWK Schriftenr 100:1–96
Schweizer G (1975) Untersuchungen zur Physiogeographie von Ostanatolien und Nordwestiran, geomorphologische, klima- und hydrogeographische Studien im Vansee- und Rezaiyehsee-Gebiet. Tübinger Geogr Studien 60:145 pp, Selbstverl. Geogr Inst Univ Tübingen
Sieger R (1888) Die Schwankungen der hocharmenischen Seen seit 1800 in Vergleichung mit einigen verwandten Erscheinungen. Mitt K K Geogr Ges 95–115(159–181):390–426
Spencer RJ, Eugster HP, Jones BF (1985) Geochemistry of Great Salt Lake, Utah II: Pleistocene-Holocene evolution. Geochim Cosmochim Acta 49:739–747. doi:10.1016/0016-7037(85)90168-1
Stumm W, Morgan JJ (1996) Aquatic chemistry, 3rd edn. Wiley, New York, pp 906–908
Takahashi T, Broecker WS, Langer S (1985) Redfield ratio based on chemical data from isopycnal surfaces. J Geophys Res 90:6907–6924. doi:10.1029/JC090iC04p06907
Tuğrul S, Dümlu G, Bastürk Ö, Ilhan R, Balkas T (1984) Van Gölü Özümleme Kapasitesinin. Saptamasi Ve Evsel Nitelikli Atiksu. Aritimi Ve Desarji Optimizasyonu. TÜBITAK Report, Proj. No. 0730018301, Van, 185 pp
Valeton I (1978) A morphological and petrological study of the terraces around Lake Van, Turkey. In: Degens ET, Kurtman F (eds) Geology of Lake Van, Miner Res Explor Inst Turkey 169, Ankara, pp 64–80
Wick L, Lemcke G, Sturm M (2003) Evidence of Lateglacial and Holocene climatic change and human impact in eastern Anatolia: high resolution pollen, charcoal, isotopic and geochemical records from the laminated sediments of Lake Van, Turkey. Holocene 13:665–675. doi:10.1191/0959683603hl653rp
Wong HK, Degens ET (1978) The bathymetry of Lake Van, eastern Turkey. In: Degens ET, Kurtman F (eds) Geology of Lake Van, Miner Res Explor Inst Turkey 169, Ankara, pp 6–10
Wong HK, Finckh P (1978) Shallow structures in Lake Van. In: Degens ET, Kurtman F (eds) Geology of Lake Van, Miner Res Explor Inst Turkey 169, Ankara, pp 20–28
Wong HK, Degens ET, Finckh P (1978) Structures in modern Lake Van sediments as revealed by 3.5 KHz high resolution profiling In: Degens ET, Kurtman F (eds) Geology of Lake Van, Miner Res Explor Inst Turkey 169, Ankara, pp 11–19
Wüest A, Imboden DM, Schurter M (1993) Vertikale Dichtestruktur und Mischung im Van See. Kap. 3 in Abschlußbericht, DFG Projekt Wo 395/2-1 bis 2-4. Inst Biogeochem Meereschem Univ Hamburg, 20 pp (unpublished)
Acknowledgments
The field research was funded by the DFG Project WO 395/2-(1-4) and by the Volkswagen Foundation. The field work would not have been possible without the devoted cooperation and friendship of the late Tosun Konuk, University of Izmir. The following colleagues helped in the field work: 1989: Prof. Dr. Tosun Konuk, Dr. Mohammat Duman, Dogan Yasar, Neyat Somali, University of Izmir; Prof. Dr. Jozef Kaźmierczak, Polish Academy of Sciences, Warshaw; Prof. Dr. Dieter Imboden, Prof. Dr. Alfred Wüest, Michael Schurter, ETH Zürich; Dr. Andreas Lipp, Dr. Uwe Svensson, University of Hamburg; 1990: Prof. Dr. Tosun Konuk, Dr. Muhammet Duman, Dr. Doğan Yaşar, Nejat Somali, University Izmir; Dr. Gerry Lemcke, Markus Hofer, Michael Schurter, ETH Zürich; Dr. Eva Danulat, Dr. Andreas Lipp, Dr. Uwe Svensson, University of Hamburg. Without their persistent help and good humor, even under difficult external conditions, the here presented results could not have been obtained. We also have to thank the crew of the tug boat Süphan, Tatvan, for mastering some difficult marine maneuvres, not within their normal schedule.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Lithology, chronology, and mineralogical-geochemical characteristics of Lake Van sediments (core K10, 420 m depth).


Sediment deposition rate (SDR), and mineralogy (relative intensities according to X-ray diffraction analysis) of core K10 plotted versus revised sediment age (Landmann and Kempe 2005). In the younger sediment sequence, the low background signal of dolomite is assumed to represent mostly detrital input from the drainage area. Occurrences of protodolomite (squares) and magnesite (triangles) together with their Ca:Mg ratios are given in the dolomite curve

Element distribution of the lithogenic and organic sediment fractions of core K10 used to calculate element deposition rates from SDR given in Fig. 11. Ccarb: carbonate carbon; Plith: inorganically bound phosphorous; Siopaline: extractable opaline silica; Corg: organic carbon; Ntot: total nitrogen; Porg: organic phosphorous
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Reimer, A., Landmann, G. & Kempe, S. Lake Van, Eastern Anatolia, Hydrochemistry and History. Aquat Geochem 15, 195–222 (2009). https://doi.org/10.1007/s10498-008-9049-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10498-008-9049-9